| Nick Shulman responded: |
2022-05-17 16:00 |
The best way to see documentation for Skyline's command line is with the menu item "Help > Documentation > Command Line".
The documentation that you can find on web pages is always going to be out of date.
The Skyline commandline does not really have a way of creating a new document. It is expected that you will use the Skyline user interface to create an empty document with the settings that you need. Then, you would use the commandline interface to tell Skyline to import results into that document, etc.
Typically you would have the .sky file for a blank document somewhere and your script would copy that file somewhere and then tell Skyline to do something with it.
By the way, we recently released a new tool called "Skyline Batch" which makes writing commandline scripts for Skyline much easier. You can download Skyline Batch from here:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=skyline-batch
-- Nick |
| |
| jingyangzhang0222 responded: |
2022-05-18 11:51 |
Thank you, Nick! I used an empty template Skyline file as suggested, and now I am trying to reproduce the steps in the DIA tutorial(https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_dia).
These are the command lines I used:
./SkylineRunner.exe
--in=PATH_TO_COPIED_EMPTY_SKYLINE_FILE
--import-search-file=PATH_TO_PEPTIDE_SEARCH_RESULT
--import-file=PATH_TO_REPLICA_1
--import-file=PATH_TO_REPLICA_2
--import-fasta=PATH_TO_FASTA_FILE
--save
Is this the correct workflow for a DIA analysis?
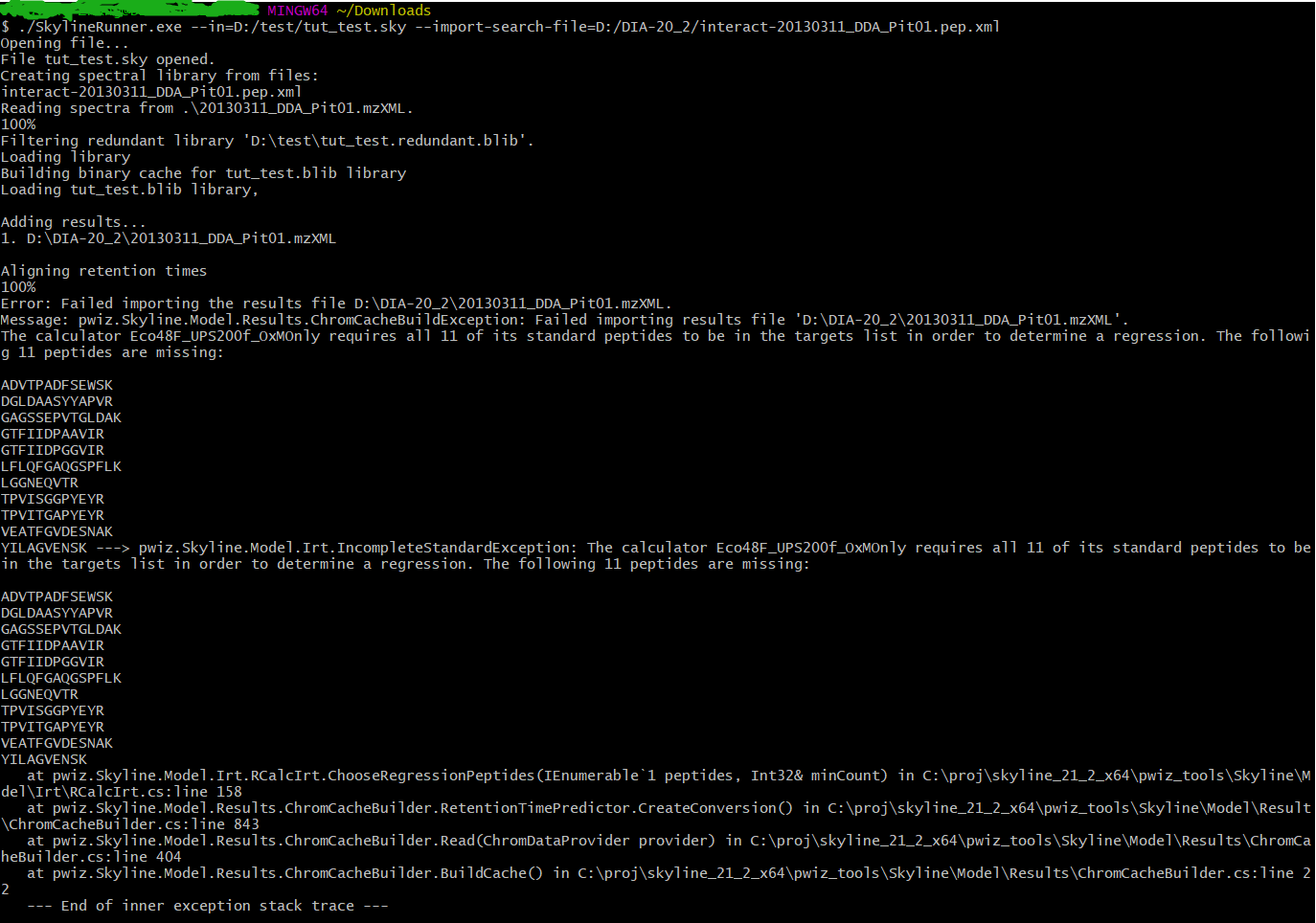
And there is an exception in the process(See the attached Image), am I setting something wrong?
|
|
| |
| Nick Shulman responded: |
2022-05-18 12:09 |
The error is saying that your Skyline document is missing one or more of the iRT standard peptides.
I am not an expert on the Skyline commandline, but it looks like maybe you need to specify "--import-search-irts" so that Skyline will know how to populate the iRT database and which iRT standard peptides to add to your document.
By the way, if you would like to learn more about iRTs you should take a look at the iRT tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_irt
-- Nick |
| |
| jingyangzhang0222 responded: |
2022-05-19 12:25 |
Hi Nick, I removed that irrelevant calculator from the empty template Skyline File, and it works!
There are the commands I am using to reproduce the result in IDA tutorial:
--in=D:/test/tut_test.sky
--import-search-file=...
--import-search-cutoff-score=0.95
--import-search-irts=None
--import-file=D:/DIA-20_2/20130311_DIA_Pit01.raw
--import-file=D:/DIA-20_2/20130311_DIA_Pit02.raw
--import-fasta=D:/DIA-20_2/pituitary_database.fasta
--save
The Skyline Document was correctly modified and saved, but there were no peaks shown, I have tried some options(like --refine-max-precursor-only) but it still did not work. Since you are not an expert on the Skyline commandline, how can it show the peaks and color dots by adjusting settings in the user interface? Pls refer to the screenshots attached.
If you know the commandline options to correct this result, it would be the best. :)
|
|
| |
| Nick Shulman responded: |
2022-05-19 12:40 |
Can you send us your Skyline document that you ended up with?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms. If that .zip file is less than 50MB you can attach it to this support request.
You can upload larger files here:
https://skyline.ms/files.url
--Nick |
| |
| jingyangzhang0222 responded: |
2022-05-19 18:38 |
|
| |
| Nick Shulman responded: |
2022-05-19 22:15 |
The document that you uploaded does not look the same as the one that you took a screenshot of in Skyline ("Skyline UI.png").
I was hoping that you would start from the document in the screenshot and do "File > Share" to create a .sky.zip file and upload that one.
The document that you uploaded has 3 replicates, six proteins, but no peptides.
If you could upload the document that you took the screenshot of, I could probably tell you why you do not see any chromatograms.
The usual reason that you end up with no chromatograms is usually that there were no spectra in the raw file which matched the precursors in your document. This usually has something to do with the settings on "Settings > Transition Settings > Full Scan".
Can you try uploading a different Skyline document? Also, it might be helpful if you could send me one of your .raw files such as "20130311_DIA_Pit01.raw".
-- Nick |
| |
| jingyangzhang0222 responded: |
2022-05-22 08:10 |
|
|
| |
| Nick Shulman responded: |
2022-05-22 09:58 |
Oops. I see now that the file you uploaded does contain peptides, precursors and transitions. What is actually happening is that as soon as I open your document in Skyline, Skyline removes all of the peptides from the document.
The reason that Skyline does this has something to do with your settings at:
Settings > Peptide Settings > Digestion
You have "Enforce uniqueness by" set to "Proteins".
You should change this to "None"
I believe that what is happening is that Skyline sees that none of the peptides in your document are unique to the proteome and the peptides get removed from the document.
I am not sure whether this peptide uniqueness constraint is what is causing you not to get the chromatograms that you want. I would recommend that you change that setting to "None" in the document that you are starting with, and then do your command line steps again. This might fix your missing chromatogram problem. If that problem is still not fixed, please upload a new .sky.zip file.
-- Nick |
| |
| jingyangzhang0222 responded: |
2022-05-23 08:18 |
Hi Nick,
I have set every option in "Peptide Settings" and "Transition Settings" the same as that in the DIA tutorail(including "Enforce uniqueness by").
After modifying the empty template Skyline File, I used the command line to copy and run SkylineRunner commands.
The result still does not show the chromatograms:( Skyline File can be found here( https://skyline.ms/_webdav/home/support/file sharing/@files/0523_tut_test.sky.zip?contentDisposition=attachment). |
| |
| Nick Shulman responded: |
2022-05-23 08:49 |
I am not an expert on the Skyline commandline but the behavior that you are seeing is what you would get if the peptides were added to the document after the chromatogram extraction happened.
I wonder whether you could try splitting this up into two separate operations:
--in=D:/test/tut_test.sky
--import-search-file=...
--import-search-cutoff-score=0.95
--import-search-irts=None
--import-fasta=D:/DIA-20_2/pituitary_database.fasta
--save
--in=D:/test/tut_test.sky
--import-file=D:/DIA-20_2/20130311_DIA_Pit01.raw
--import-file=D:/DIA-20_2/20130311_DIA_Pit02.raw
--save
-- Nick
|
| |
 Exception.png
Exception.png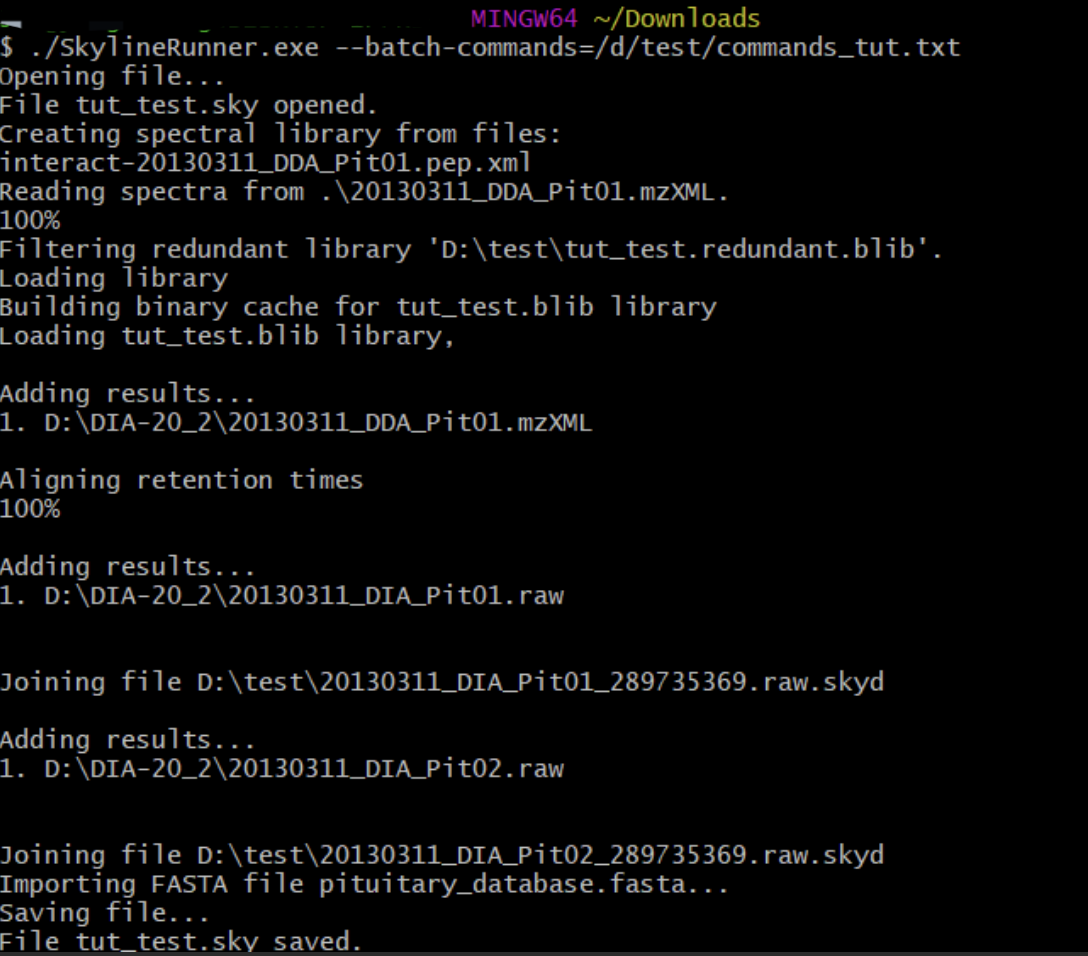 CommandLine.png
CommandLine.png Skyline UI.png
Skyline UI.png screenshot_0522_commandline_UI.png
screenshot_0522_commandline_UI.png