| How to visualize and export y and b ions from M+1 and M+2 precursors | erints | 2026-03-16 15:44 | |||||||||||||||||||||||||||
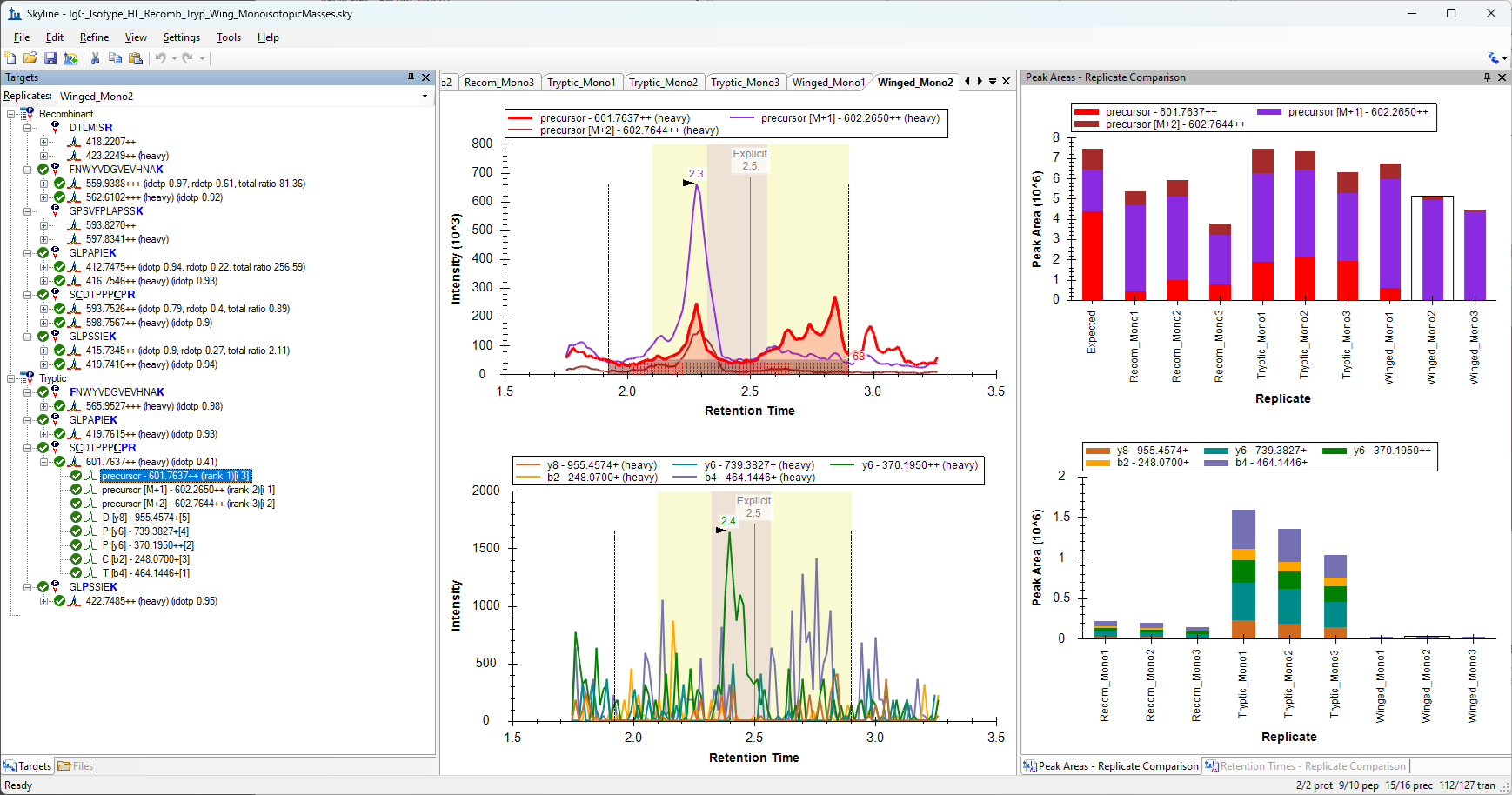
Hello, We are looking to resolve some interference we're having between several heavy-labeled internal standard types we are interested in comparing. We are looking at IgG isotype peptides and only have this interference occurring with our IgG3 peptide. One way we are looking at resolving this, is by looking at the M+1 or M+2 precursor for select standards instead of the monoisotopic precursor to increase the distance between the masses, especially since the tryptic IgG3 standard has higher signal for the M+1 over monoisotopic. We have already compared the monoisotopic vs average, but this didn't resolve the issue for our specific isotype of interest. Is there a way where we can change a setting in Skyline or use a reporting feature so we can isolate what the product m/z is for the M+1 and M+2 precursors for a specific peptide? I can see the M, M+1 and M+2 m/z precursors in the pulldown, but the monoisotopic precursor is what is used to visualize/quantitate the respective product's m/z. I tried only checking the M+1 and reimporting, thinking that Skyline would recalculate the m/z for the products just for the M+1 precursor, but it continues to give products for the monoisotopic precursor only. We are looking at how to find these values in prospector, but it would be great if there was already a feature in Skyline that would display products for a specific precursor, especially since those precursor values are already known. We are currently using Skyline 26.1. Here is a screenshot of the precursor I am interested in, in a word document. Attached is the skyline file as well. Thank you! |
|||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
 MissingMonoisotopicArea.png
MissingMonoisotopicArea.png