| accuracy of light and heavy precursors differing by 1 Da only | rameshkr | 2025-02-28 04:02 | |||||||||||||||||||||||||
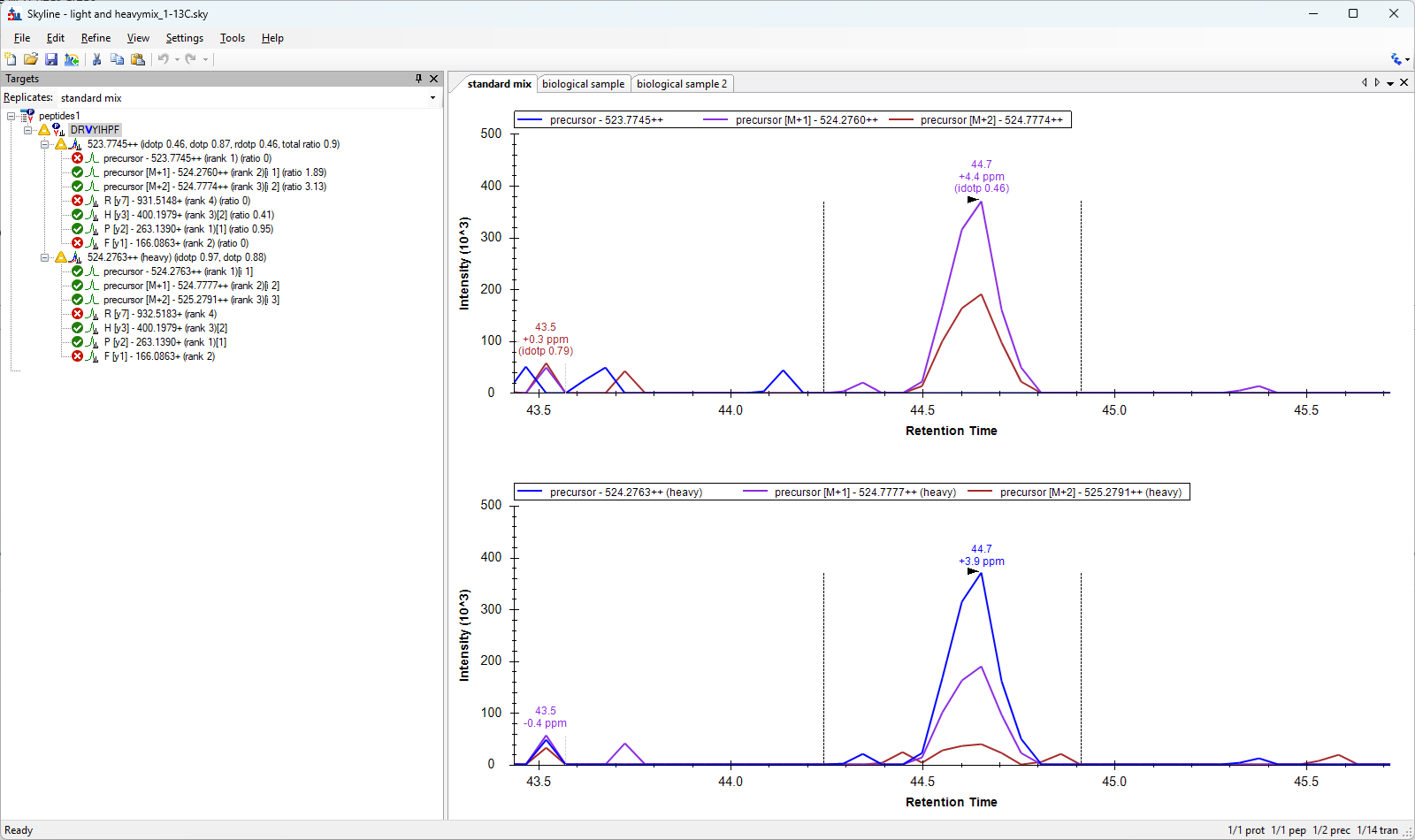
Hi team I have two peptides light and heavy (8 amino acids long). The heavy peptide has a single 13C on the 3rd amino acid so differs from light peptide by 1 Da only. I want to spike the heavy peptide in the light and monitor their +2 charge precursors (m/z difference 0.5 only). I have generated a skyline document and it gives me details for both but I was wondering how to verify that the peaks are being correctly identified- I am getting same product ions for both. Also, the M+1 and M+2 of light is same (upto 3 decimal places) as M and M+1 of heavy. I am acquiring data on Thermo Q Exactive Plus in PRM mode using 1.6 m/z isolation window and 0.5 m/z offset. I have set skyline ion and method match tolerance to 0.01 m/z under the transition settings. I have shared the skyline document and raw files on the portal (file name: light and heavy mix_1-13C). It has 3 files, standard- mix of purified light and heavy peptide and biological- two technical replicates of heavy peptide spiked in a tissue peptide extract. The results of all three are confusing. My aim is to normalize peak intensity/areas of light with heavy. How should I do that? Thanks |
|||||||||||||||||||||||||||
| |||||||||||||||||||||||||||
 Ms1Chromatograms.png
Ms1Chromatograms.png