It looks like your peptide search engine matched spectra to "ALLNAIGNLELTGAFAEALK" in several of your replicate.
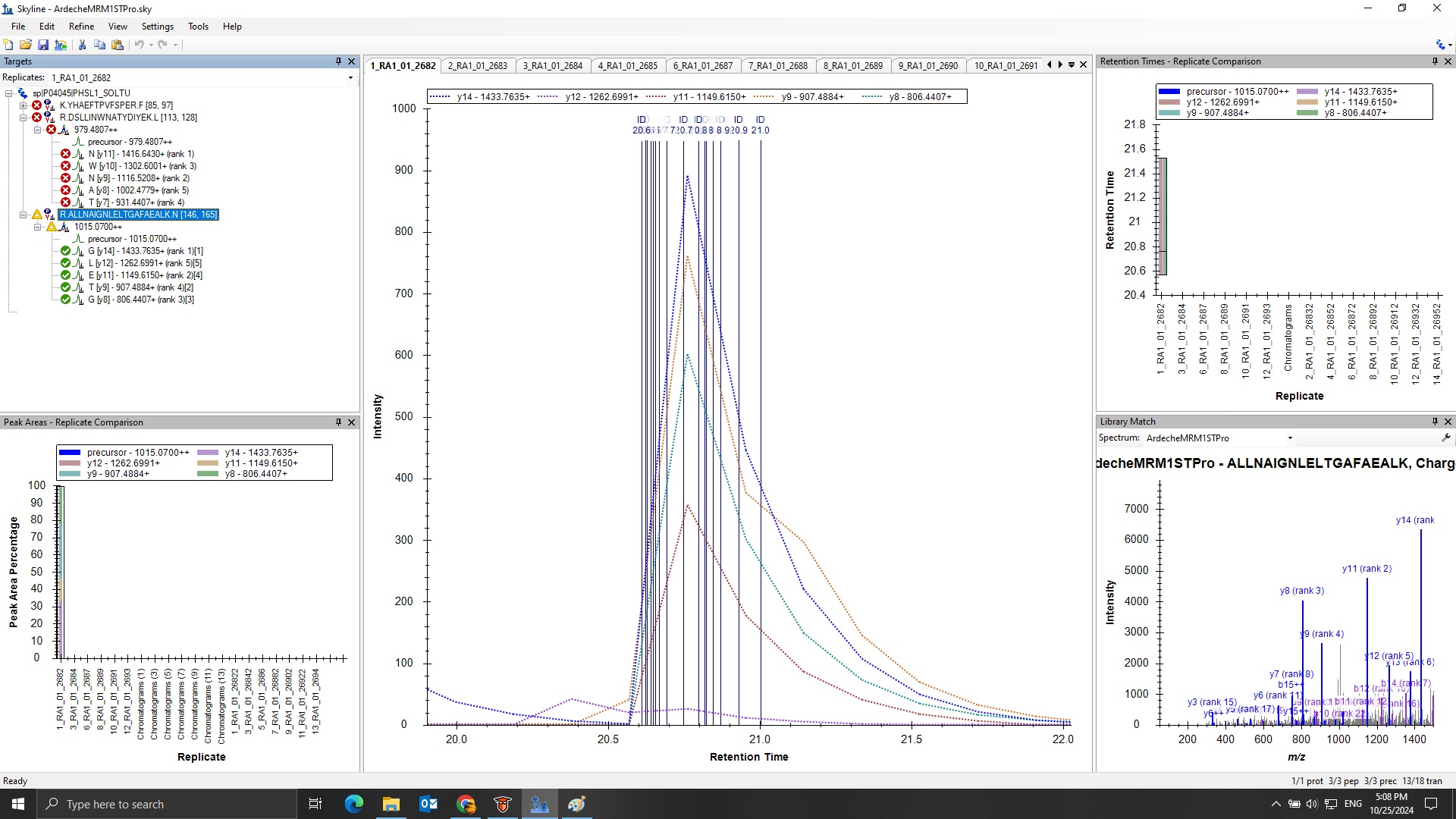
In your first screenshot, Skyline is drawing dozens of ID lines on the chromatogram graph, which means that there were many peptide spectrum matches (PSMs) for that peptide in that file.
In your second screenshot, there is a single ID line which means that there was one PSM for that peptide in that file.
The reason that the ID line is red in that second screenshot is because that is the spectrum which is being shown in the "Library Match" window.
There is a dropdown at the top of the Library Match window where you could choose a spectrum from a different replicate, and, if you chose something else the color of that ID line would change.
I do not understand what you mean when you say "none of the other peptides in this sample are detected, and this same peptide isn't identified in the other replicates". Every peptide has been detected in several replicates.
If you want to see the complete list of peptides you can go to:
View > Spectral Libraries
If you select a peptide in the Library Explorer, you can use the "File" dropdown below the spectrum to see which other files the peptide was detected in.
I see that at "Settings > Transition Settings > Full Scan" you have the MS/MS Acquisition Method set to "DDA".
When Skyline extracts chromatograms from DDA data, the chromatograms usually look very strange with long straight lines connecting the places where a matching precursor happens to have been selected by the mass spectrometer for fragmentation. For this reason, DDA MS2 chromatograms in Skyline are not useful for quantification and, so, Skyline displays the MS2 chromatograms as dotted lines.
Another thing that you might have noticed is that if you do not also tell Skyline to extract MS1 chromatograms in addition to the MS2 chromatograms, you do not get any MS2 DDA chromatograms. It looks like you might have already worked around this by setting the "MS1 Isotope peaks included" at "Settings > Transition Settings > Full Scan" to "Count".
Skyline is primarily a tool for extracting chromatograms. It is possible to use Skyline to look at DDA peptide search results, but the features that Skyline has in this area only exist because they are also useful for extracting chromatograms. If all you want to do is look at your DDA search results, there are probably better tools, but I am not sure what they are.
I see that your spectral library was created from .mzid files, so I am guessing the peptide search engine that you used was MSGF+, but it might have been something else.
You might find some helpful information in one of Skyline's tutorials in the "Full-Scan Acquisition Data" section:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorials
-- Nick
 1.jpg
1.jpg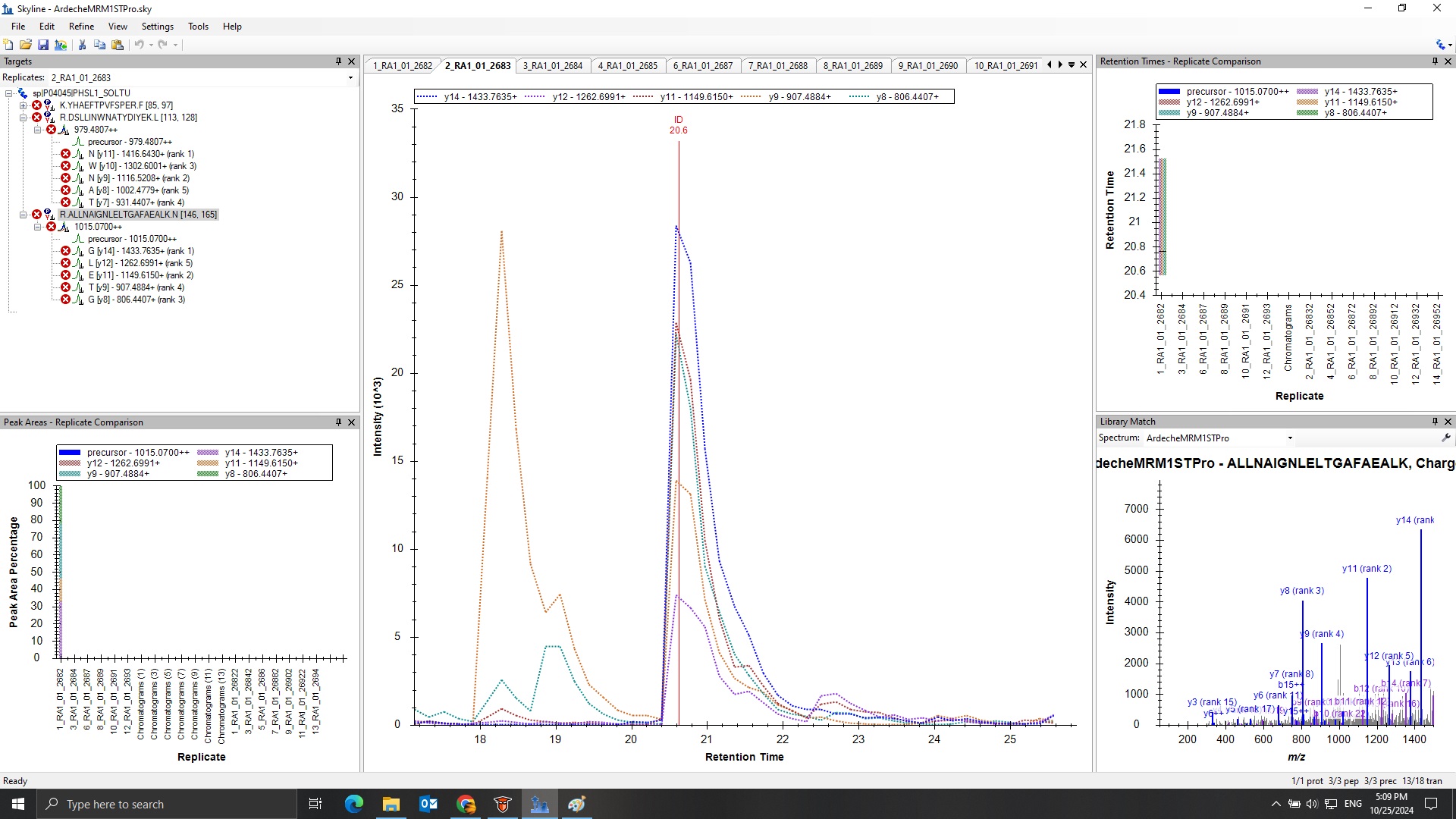 2.jpg
2.jpg msconvert_Setting.jpg
msconvert_Setting.jpg