| Defining retention time for peptide quantification | mt | 2023-03-28 02:20 | |||||||||||||||||||||||||||||||||||||||||
Hi, I started using Skyline (version 22.2.0.351) recently and have been trying to set up a peptide peak quantification protocol but I have trouble effectively defining retention time windows for peak detection. I managed to define the Explicit RT and Explicit RT Window in the Document Grid, but the issue I keep having is that the right peak is picked only if Explicit retention time falls exactly within a peak. More precisely, it has to fall within a peak integration boundaries that Skyline does defines every time, but are presented as either black or grey dashed lines, depending on the picking success. If the defined explicit retention time is outside of these boundaries, random noise is picked near the defined retention time, even though the actual peak might be eluting just a few seconds before or after the explicit RT and is well within the RT window. Re-scoring and re-importing doesn't help. Thank you! |
|||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||
 Screenshot 2023-03-28 at 11.05.37.png
Screenshot 2023-03-28 at 11.05.37.png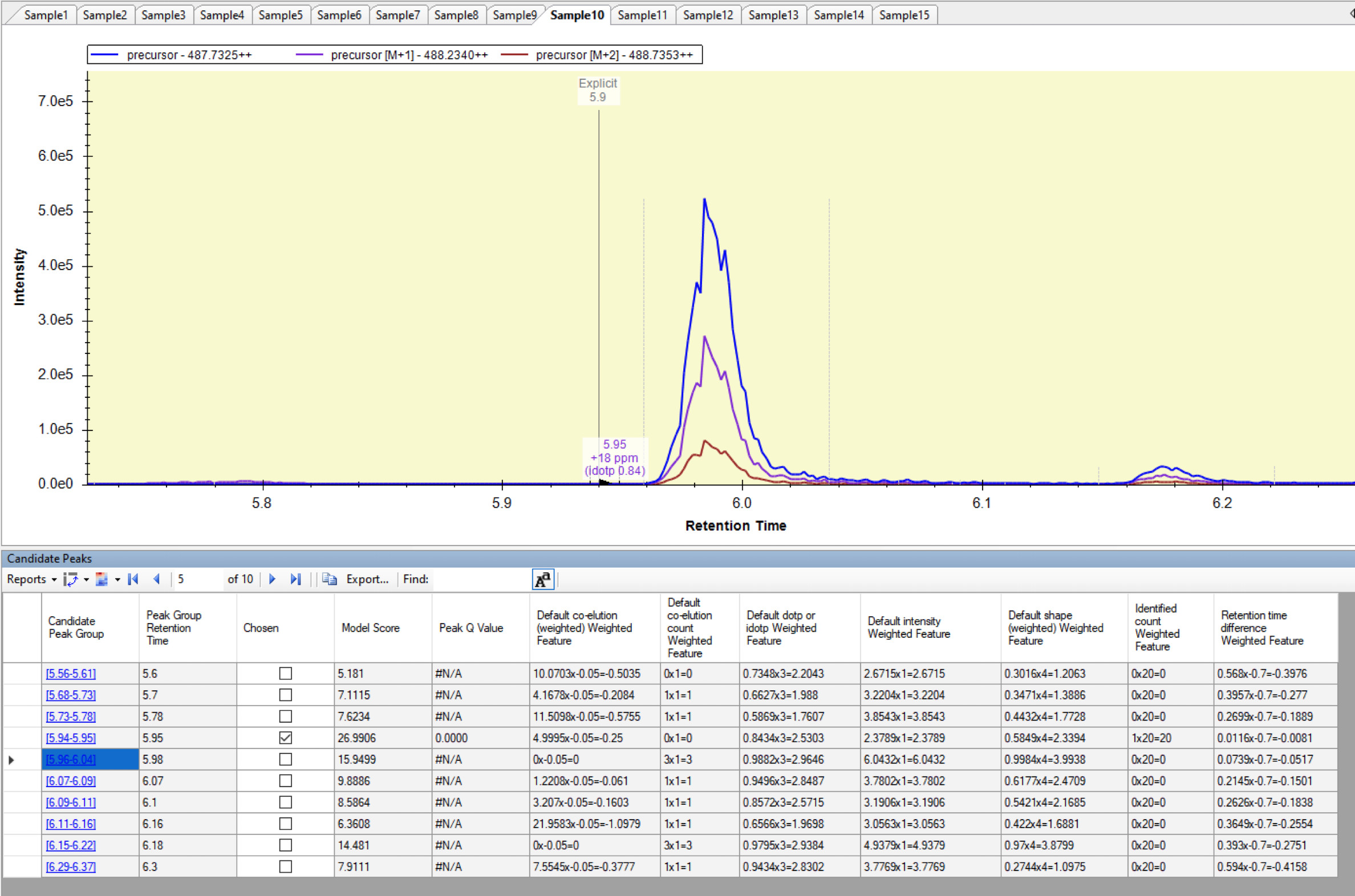 Screenshot_Sample10.png
Screenshot_Sample10.png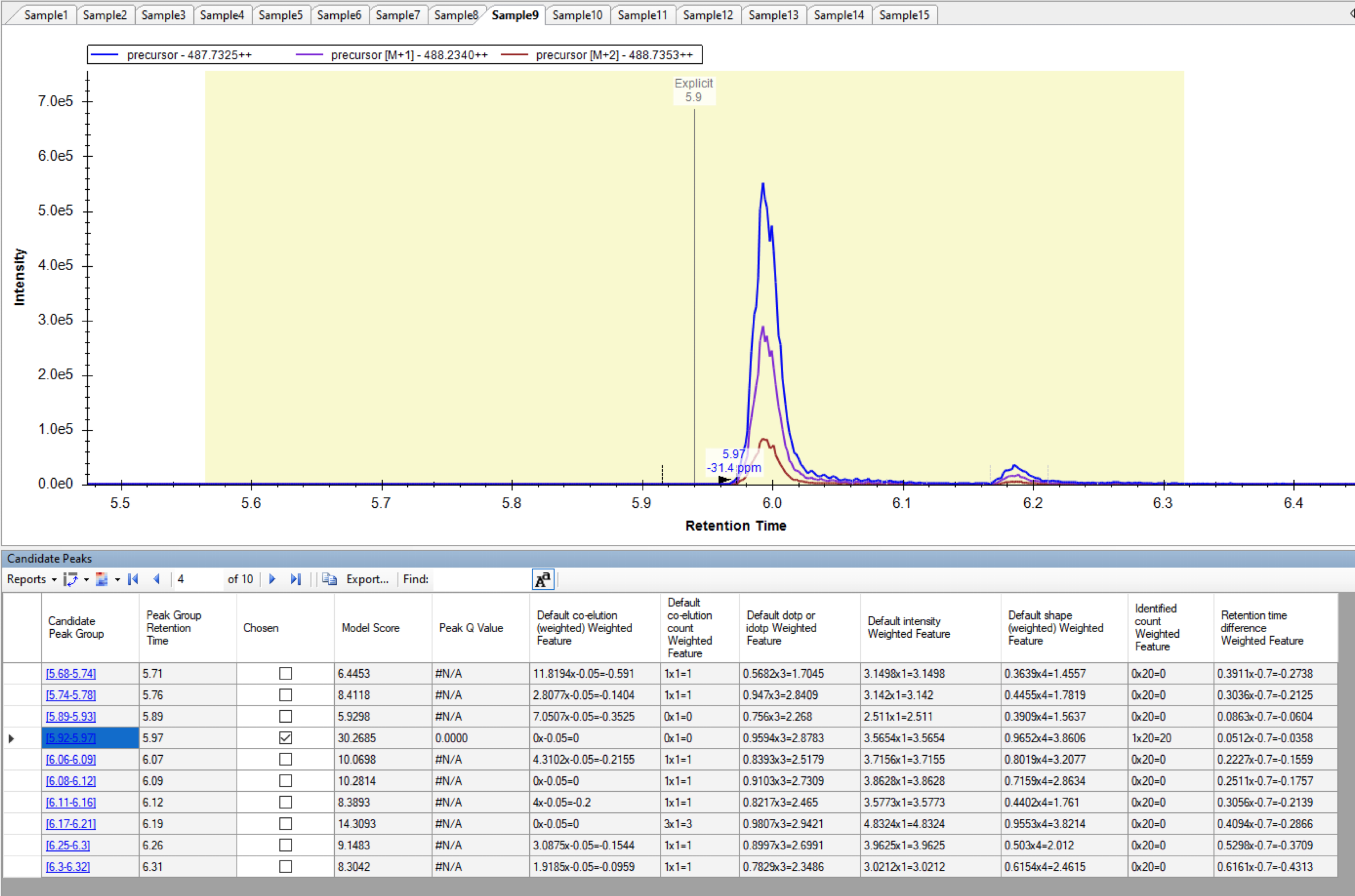 Screenshot_Sample9.png
Screenshot_Sample9.png