Edit > Insert > Protein
would use whatever enzyme you have selected at "Settings > Peptide Settings > Digestion". That defaults to being Trypsin.
You would only see something in the Library Match window if you had a spectral library. You can also right-click on the Library Match window and choose "Prosit" which means that instead of Skyline showing you a spectrum from your spectrum library, Skyline instead will ask a server in Germany to use its machine learning algorithm to predict the fragmentation pattern of the currently selected precursor.
(If you still don't see anything in the Library Match window after selecting "Prosit", you probably don't have any peptide or precursor in your document. You should send me your Skyline document (see answer #2 below))
1) The spectral library tells Skyline which fragment ions are likely to be detected for any given peptide.
1.1) Skyline does not know how to download FASTA files from anywhere, so you should download it yourself.
2) Can you send us your FASTA file and your Skyline document?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files such as spectral libraries
If that .zip file and your FASTA file are less than 50MB you can attach them to this support request.
You can upload larger files here:
https://skyline.ms/files.url
There are lots of potential reasons that Skyline might decide not to add any of the proteins from your FASTA file to your document and after I see your FASTA file and your .sky.zip I will be able to tell you what is going wrong.
3) You create a background proteome by going to "Settings > Peptide Settings > Digestion" and choosing "<Add...>" from the Background proteome dropdown. If you are just starting out with Skyline I would discourage you from choosing anything other than "None" in the "Enforce peptide uniqueness" since Skyline automatically removing non-unique peptides from your document sometimes results in confusing behavior.
4) "[KR | P]" indicates that the enzyme cleaves at Lysine or Arginine unless that amino acid is followed by a Proline. If you want to see the details of the currently selected enzyme you can go to:
Settings > Peptide Settings > Digestion
and choose "<Edit current...>" from the Enzyme dropdown and Skyline will show you a dialog with all of the details about the currently selected enzyme.
-- Nick
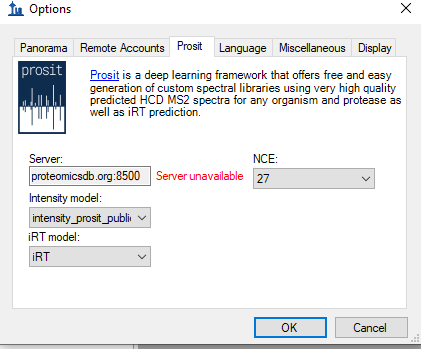 Prosit server failure.PNG
Prosit server failure.PNG peptide atlas cannot import into skyline query.PNG
peptide atlas cannot import into skyline query.PNG