| Rugged extracted ion chromogram of targeted peptide precursors from FAIMS-DIA file by Skyline. | Winnie | 2022-10-30 07:38 | |||||||||||||||||||||||||||||||||
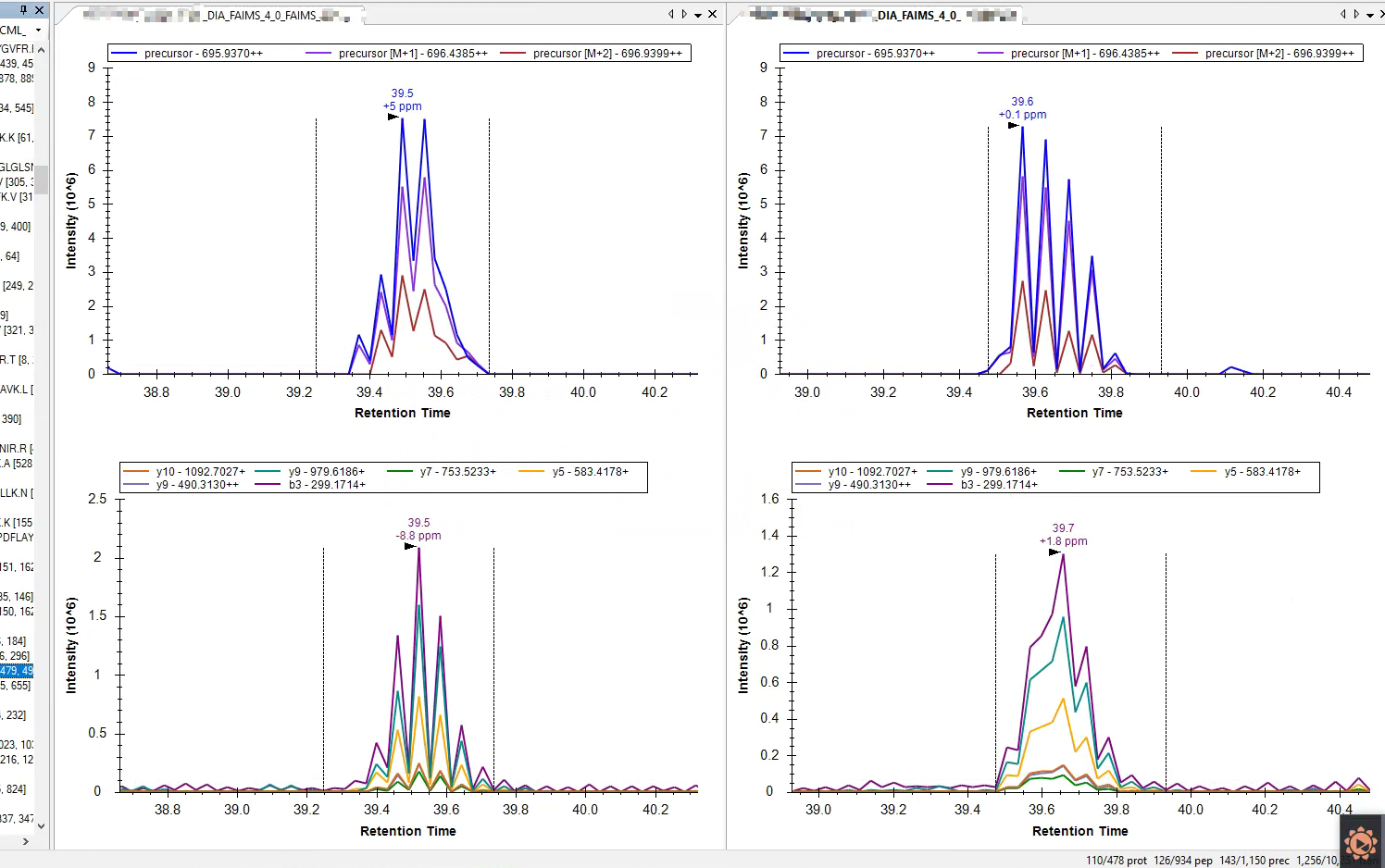
Hi, skyline, I extracted chromotogram of targeted peptide precursors from FAIMS-DIA files by skyline, the xic show rugged profiling. The screenshot is attached. I was wondering there are issue on ion moblity library because i did not set ion mobility sepctral library. The sepctral library was imported by the following strategy. File>import>assay library. I found the column named precursor ion mobility in this spectral library is NA. The spectral library was build by Fragpipe with files acquired by FAIMS-DDA. I first communicated with Fragpipe team, they told me it is normal the column named precursor ion mobility is NA, because they this the FAIMS is low resolution device for ion moblity. So they set NA of precursor ion mobility for the library genetated by FAIMS-DDA. So, did you have suggestion on the rugged xic and how to spectral library generated by FAIMS-DDA |
|||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||
 Screenshot.png
Screenshot.png