| |
| Nick Shulman responded: |
2022-05-23 09:14 |
There are some ion mobility related tutorials on this page:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_ims
Let us know where you are getting stuck and we can point you in the right direction.
Or, if you do not know how to get started, let us know what sort of data you have. Do you have a list of molecules and transitions you would like to measure? Do you know what their ion mobility values are, or are you going to want Skyline to figure that out by looking at the raw data you collected?
If you have some files that you would like to send us, you can attach files which are less than 50MB to this support request. You can upload larger files here:
https://skyline.ms/files.url
-- Nick |
| |
| Juan C. Rojas E. responded: |
2022-05-24 01:12 |
Hi F. J. Diaz-Galiano and Nick,
So for sure the Skyline team can help you better, but in case it might be helpful there is a document that I prepared precisely to introduce Waters users into how to exploit Skyline.
You can actually find some example data and a detailed tutorial for data analysis of bottom-up proteomics data in:
https://skyline.ms/project/home/support/file%20sharing/begin.view?
In the folder "Day_1_am_14_00_60min_Rojas_Session4"
I hope this helps.
Sincerely,
Juan C. Rojas E. |
| |
| diaz-galiano responded: |
2022-05-24 01:15 |
Hello, Nick,
Thank you so much for your prompt reply. I had reviewed that tutorial but did not know how to apply it to my own files, or how to get started in general.
We have HDMS data from a Synapt G2-Si (ion mobility, elution time and full scan MS only) of 24 compounds. The ion mobility technique is TWIMS, so we have a calibration curve (mob_cal.csv file in the sample root) that the proprietary software uses to correlate measured drift times to CCS values. We would also like to integrate the chromatograms for quantitation purposes. ** I forgot to mention that we are performing target analysis, and that the m/z values and CCS values are already known.**
We have also tried using MSConvertGUI from the ProteoWizard Tools to convert the .raw folders into Skyline files, but cannot seem to locate any of the expected .sky extensions in this software, and thus haven't managed to open our data on Skyline. Is this the correct tool?
We have uploaded one of our HDMS samples (220520007.raw folder compressed as a .zip file) to the suggested URL.
Best regards,
Edit:
Thank you so much for the linked tutorial, Juan C. Rojas, your answer came up while I was writing this response to Nick. We are still unsure on how to proceed with Waters HDMS data conversion at this point, however, is there a tutorial on that which we could follow? Best regards,
|
| |
| Nick Shulman responded: |
2022-05-24 06:51 |
Here is the .pdf file in the group of files that Juan C. uploaded:
https://skyline.ms/_webdav/home/support/file%20sharing/%40files/Day_1_am_14_00_60min_Rojas_Session4/IMSTrainingWaters_DIA.pdf
The link to the whole folder is this:
https://skyline.ms/_webdav/home/support/file%20sharing/%40files/Day_1_am_14_00_60min_Rojas_Session4/
You do not need to convert your raw data before you use it in Skyline. Skyline knows how to read all of the same file formats as ProteoWizard MSConvert understands.
The menu item to tell Skyline to extract chromatograms from raw data is:
File > Import > Results
You will need to do a few things in Skyline before you can extract chromatograms. Those things are:
1. Tell Skyline which molecules you want to monitor. For small molecules, this is typically done by importing a transition list with the menu item:
File > Import > Transition List
Skyline will usually need to bring up a dialog which lets you tell Skyline how the columns in your transition list map to the data that Skyline is expecting.
2. Tell Skyline how to extract chromatograms from your full scan data.
It looks like your file only has MS1 data, so you should go to:
Settings > Transition Settings > Full Scan
and change "MS1 filtering Isotope peaks included" to something, probably "count".
3. Now you are ready to tell Skyline to extract chromatograms from your data. Use the menu item:
File > Import > Results
to point Skyline at your .raw data folder.
The chromatograms will probably look lousy because no ion mobility filtering was done.
4. To add some ion mobility information, go to:
Settings > Transition Settings > Ion Mobility
and choose "Add" from the "Ion mobility library" dropdown.
5. In the Edit Ion Mobility Library dialog, push the "Create" button to create a .imsdb file on disk which will hold your ion mobility information. Then, push the "Use Results" button to tell Skyline to look at your chromatograms and decide for each molecule what the best ion mobility value is.
It sounds like you already know what the ion mobility values are supposed to be for your molecules. That probably means you don't need to use the "Use Results" button to tell Skyline to choose ion mobility numbers. There is probably something else you are supposed to do, but I do not know what that is. The Skyline ion mobility expert is out of town this week but hopefully this will be enough to get you started.
-- Nick |
| |
| diaz-galiano responded: |
2022-05-25 03:27 |
Thank you for the instructions, we have managed to create a transition list and import it into Skyline. However, when we try to import a sample (the same we uploaded to the support platform), Skyline crashes with no output error.
Attached are a screen capture of the Transition settings we are using (we have tried the different options regarding retention time at the bottom) and our transition list in .csv format (molecule name, formula, precursor m/z, retention time, retention time window, adduct, charge).
We have tried selecting different options on the settings screens but to no avail. We are working in sensitivity mode (no high mass accuracy) and haven't been using lock mass, in case it is related somehow to our impossibility to import the results.
Best regards,
Edit: We have reproduced this situation on three different computers (U.S. English, Spanish and French system languages).
Edit 2: We have managed to open a file, however, it was an HRMS file (without ion mobility experiments, i.e. not an HDMS file). Apparently, Skyline is having issues opening our HDMS files at the moment, perhaps because we don't have an ion mobility database. Thus, we are unable to create an ion mobility library based on our results. We are going to try to create one looking at examples and/or tutorials, and see if we manage to open an HDMS (or follow any other instructions you could provide).
We are very thankful for all of the support team (and Juan's) time, we know we have been bombarding you with questions for the last few days. |
|
| |
| Nick Shulman responded: |
2022-05-25 07:09 |
What do you mean "crashes with no output error"? What do things look like when that happens? Could you send us a screenshot?
Can you send us your Skyline document?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including whatever chromatograms you might have manage to extract.
After I receive your Skyline document I will try extracting chromatograms (File > Import > Results) from the .raw folder you have already sent us, and see what might be going wrong.
-- Nick |
| |
| diaz-galiano responded: |
2022-05-27 01:08 |
As soon as I try to import the results into Skyline, a window with the title "Importing results" closes, as does Skyline itself. I have included a screenshot of the window that closes down taken on a different file (HRMS, not HDMS), I cannot take a picture of the process of crashing because it is instantaneous.
When I use our Skyline transition list (attached to this post) to open a Waters .raw file without ion mobility (just a high resolution, full scan analysis), it opens and imports the results just fine. It only crashes on the files that have ion mobility data.
We are having crashing issues on different computers with the raw file I updated and with the Skyline transition list attached.
Best regards, |
|
| |
| Nick Shulman responded: |
2022-05-27 07:17 |
Thank you for sending that Skyline document.
Yes, Skyline crashes on my computer too, disappearing without any sort of error message.
The behavior that you are seeing looks exactly the same as what was reported by another user this week:
https://skyline.ms/announcements/home/support/thread.view?rowId=55847
I am not sure whether the crash that you are seeing is being caused by the same bug as this other user. I will ask around and make sure that someone fixes whatever bug is causing Skyline to disappear when trying to extract chromatograms from your data file.
-- Nick |
| |
| diaz-galiano responded: |
2022-05-30 00:52 |
I have read that support link, thank you for providing it, Nick. It seems this person is trying to extract results from an MRM file from a Xevo G2-XS, which a quick Google search indicates it's also a QTOF. I don't know if they have Full Scan data alongside MRM data on their file, and having more than one experiment in the same file could be leading to the crash.
In the meantime, I have uploaded a file to the repository which does not lead to a crash on our computers (220517008.raw). It contains Full Scan data only (instead of Full Scan and Ion Mobility data), in case it may be useful for you to compare with the file that does lead to a crash.
If we may be of further assistance for anything, please, do not hesitate to contact us. Thank you for your help. |
| |
| diaz-galiano responded: |
2022-06-03 02:00 |
Hello again,
We have continued working on trying to import HDMS results into Skyline.
We have managed to import the data after using ProteoWizard version 3.0.22152 64 bit, the latest at the time of writing. It is our first time using this software, so we are not completely sure on how to better do this conversion and we are still working on it. So far, this command has been creating files that we can import as results in Skyline:
msconvert.exe INPUTFILE.raw -o OUTPUTFOLDER --mzML --filter "peakPicking true cwt 1" --filter "scanEvent 1" --combineIonMobilitySpectra
This is working for us for analysis on a SYNAPT G2-Si in full scan mode only (no MS2), without lockspray, and with ion mobility enabled.
Unfortunately, our chromatograms are not looking very good at the moment (see pictures attached). We believe it is either (a) some filter we are using in the conversion process or (b) due to the high mass error in sensitivity acquisition mode, Skyline is not detecting all mass spectra as pertaining to the target mass, and thus is not including them in the drawn chromatogram resulting in poor peak shape.
The good news is that we are able to view the drift times, at least, after having followed the ion mobility tutorial, and also have built a library of PFAS CCS and drift time values to have as a reference.
However, since we are using traveling wave ion mobility, we typically employ a CCS calibration curve (mob_cal.csv) to transform drift times into CCS values, using a calibration mixture (CCS Major Mix, by Waters).
At the moment, we are struggling on how to apply this calibration curve to a sample to obtain CCS values as we do on DriftScope to obtain the CCS values, i.e., how to use Skyline to extract CCS values from drift times using a calibration.
I hope the information on msconvert allowing to open the files in Skyline can shed some light into the direct import issue ourselves and other users were having.
We can also upload the converted files (3.5 GB) if they would be of interest to you.
Regards, |
|
| |
| Nick Shulman responded: |
2022-06-03 05:17 |
Skyline extracts chromatograms by summing the intensities across a m/z range around the predicted m/z of your molecule.
If you would Skyline to sum across a wider m/z range, you should change the Resolution (or "Resolving Power" or "Mass Accuracy", depending on what is selected as the "Mass Analyzer") at:
Settings > Transition Settings > Full Scan
I am not sure about the answers to your other questions. It might be helpful if you could send us your Skyline document ("File > Share") and one of your raw files.
-- Nick |
| |
| Brian Pratt responded: |
2022-06-03 09:29 |
The DT/CCS calibration isn't something that can be represented in mzML, so Skyline needs the original raw file for that functionality. That information is embedded in the raw file, and the necessary calculations are performed in the DLL that Waters provides.
I'm a bit busy preparing for ASMS next week but I will try to find some time to understand why Skyline is having trouble with your file.
I've come in a bit late to the discussion - have the raw file and accompanying Skyline document been uploaded? If so, by what names?
Best regards,
Brian Pratt |
| |
| diaz-galiano responded: |
2022-06-03 11:41 |
Hello, Nick and Brian,
Thank you both for your time and answers. We have managed to improve peak shapes by lowering the resolving power, that was really helpful.
It is unfortunate that mzML cannot perform that calculation, we were moderately happy with being able to import the files and view the drift times (albeit not managing to export them by any means).
Regarding Brian's question, we have uploaded the following files:
- 2205020007.raw file (zipped) containing Full Scan MS data and Ion Mobility data, without lockspray. The mob_cal.csv file for CCS calibration is included within the .raw folder. This causes a crash when trying to import the sample into Skyline (tested on stable and daily versions).
- 220517008.raw file (zipped) containing Full Scan MS data, without lockspray. This file can be correctly imported into Skyline and does not cause a crash.
- In this reply, we have included the latest .sky file (created using the daily 64 bit Skyline version). We have checked before uploading that the behaviour still takes place: non IM files are correctly imported, IM files cause a crash. The original .sky files are uploaded within previous replies to this support thread ("sky_files.zip").
Do not hesitate to ask for anything else you may need from our side.
Best regards,
PS: I wish you the best for next week's ASMS meeting, the head of my laboratory will be assisting, too, I hope I can assist another time! |
|
| |
| Brian Pratt responded: |
2022-06-14 17:11 |
Can you tell me a little more about 2205020007.raw (the one with IM data)? Specifically, about the nature of each Function? Function 2 in particular.
Thanks,
Brian |
| |
| diaz-galiano responded: |
2022-06-15 01:56 |
Hello, Brian,
I am not an expert on the insides of the files, but I have been looking around the system's software and through other previously acquired data and I have found the following information:
1. In the Instrument Setup software, there is a tab called "Functions" that allows adding an experiment to the MS setup file. The possible experiments are (1. HRMS) MS, MS/MS, PID Product, PID Neutral, MSe Continuum, MSe Centroid, Tof-MRM, Fast DDA; (2. HDMS) HDMS, HDMS/MS, HDMSe, TAP, HD-MRM, HD-DDA.
2. Whenever working on HRMS mode only (i.e. full scan and no ion mobility) and without LockSpray, files contain one function only. Both on the "_HEADER.txt" file and on the "_extern.inf" file there is only reference to one function, Function 1. The name of this function, as can be seen on "_extern.inf", is: "Function 1 - TOF MS FUNCTION" (see attached files "_HEADER_HRMS_HR.txt" and "_extern_HRMS_HR.txt").
3. Whenever working on HRMS mode only (i.e. full scan and no ion mobility) and with LockSpray, files contain two functions. Both on the "_HEADER.txt" file and on the "_extern.inf" file there is a reference to both, Function 1 and Function 2. The name of these functions, as can be seen on "_extern.inf", is: "Function 1 - TOF MS FUNCTION" and "Function 2 - REFERENCE". This "REFERENCE" function contains information about the LockSpray configuration (see attached files "_HEADER_HRMS_LockSpray_HR.txt" and "_extern_HRMS_LockSpray_HR.txt").
4. Whenever working on HDMS mode (i.e. full scan with ion mobility) and without LockSpray, files contain two functions. On the "_HEADER.txt" file, there is reference to both functions, but as far as I can tell, the information "_HEADER.txt'' contains about both functions is identical. On the other hand, "_extern.inf" contains information about only one function still, Function 1. The name of this function, as can also be seen on the relevant "_extern.inf" file, is: "Function 1 - MOBILITY MS FUNCTION" (see attached files "_HEADER_HDMS_HR.txt" and "_extern_HDMS_HR.txt").
5. If the instrument is set up for HDMS (i.e. full scan with ion mobility) and with LockSpray, files contain four functions. On the "_HEADER.txt" file, there is reference to all four functions, but again, as far as I can tell, the information "_HEADER.txt" contains about all of them is also identical this time. As for the "_extern.inf" file, now it contains information about two functions, Function 1 and Function 2. The name of Function 1 is "Function 1 - MOBILITY MS FUNCTION", while the name of Function 2 is "Function 2 - REFERENCE" (see attached files "_HEADER_HDMS_LockSpray_HR.txt" and "_extern_HDMS_LockSpray_HR.inf").
6. Comparing analyses using HRMS with LockSpray (two functions, both shown on "_extern.inf"), HDMS without Lockspray (two functions, one shown on "_extern.inf") and HDMS with LockSpray (four functions, two shown on "_extern.inf"), I have been able to identify the MS data and the IM data on the HDMS-LockSpray file by comparing relative file sizes:
6.1. _FUNC001.dat contains MS data in all HRMS-LS, HDMS-noLS and HDMS-LS.
6.2. _FUNC002.dat contains LockSpray data on HRMS-LS and HDMS-LS.
6.3. _FUNC002.dat contains IM data for the MS function on HDMS-noLS.
6.4. _FUNC003.dat contains IM data for the MS function on HDMS-LS.
6.5. _FUNC004.dat contains IM data for the LS function on HDMS-LS.
7. I have been looking through all the help guides contained in the Waters software and have not been able to locate further information on these functions. In my opinion, enabling the ion mobility function creates a second function for each scan type the user has in the instrument setup, e.g., if a user has a full scan with LockSpray setup (functions 1 and 2), it will create another set of functions for each experiment (function 3 with IM for function 1, and function 4 with IM for function 2). I have not been able to gather documental evidence to support this besides my own observations.
8. The file I uploaded named "220517008.raw" is an HRMS file without LockSpray (HRMS-noLS) that contains only one function.
9. The file I uploaded named "220522005.raw" is an HDMS file without LockSpray (HDMS-noLS) that contains two functions.
10. I can upload another HDMS file with LockSpray (HDMS-LS) containing four functions and/or an HRMS file with LockSpray (HRMS-LS) containing two functions at your request, for you to have the complete set of raw files.
I hope this is not too convoluted and that it can shed some light on this issue.
As always, do let me know if there is anything else I can do.
Best regards, |
|
| |
| Brian Pratt responded: |
2022-06-15 10:48 |
Very helpful, thanks! I think the issue is that we're not reliably identifying the lockspray function, and thus getting a crash in the Waters DLL when we ask to read it as if it were normal data.
In your point #6, you mention FUNC002 twice - is that a typo? But in general do you mean to say that FUNC001 contains the union of the other non-lockspray functions?
I'd be interested in seeing the data sets mentioned in your point #10, with a description of what each function represents.
Brian |
| |
| Brian Pratt responded: |
2022-06-15 13:43 |
As I dig into this, there is one other possibility that we should consider - the file may simply be corrupt.
Do you have any other means of viewing the data? In particular I suspect that Function 2 may involve a corrupted file. |
| |
| diaz-galiano responded: |
2022-06-15 15:09 |
Hello, Brian,
No, in point #6 that is correct. Function 1, in our case, always contains MS data, however, Function 2 may contain LockSpray or IM data depending on the experiment setup. From what I have seen on the different acquired files:
A. If you only perform an HRMS analysis (e.g., a full scan analysis), the Synapt creates a file containing function 1 only, and that will have MS data. This function 1 for MS data weighs a few hundred megabytes.
B. If you perform an HRMS analysis with LockSpray, function 1 is the MS and function 2 is the LockSpray. Data on said function 2 is registered in the _extern.inf datafile. This function 2 for LockSpray data weighs a few megabytes.
C. If you perform an HDMS analysis, then, again, function 1 contains MS data, and only this information is recorded on the _extern.inf datafile. There is also function 2, not registered in the _extern.inf datafile, containing IM data corresponding to function 1 (MS). This function 2 weighs a few dozen megabytes.
D. Finally, if you perform an HDMS analysis with LockSpray, then function 1 and function 2 as in case B are created (few hundred megabytes for function 1 and few megabytes for function 2, respectively). This two functions are recorded on the _extern.inf file. However, two additional functions are created, function 3 and function 4. Function 3 weighs a few dozen megabytes, so that matches what function 2 contains in case C, and the only remaining possibility for function 4 (weighing a few hundred kilobytes) is that it must contain IM data for LockSpray MS, function 2 in this file.
Although file size is determined by the instrument resolution and the particular sample analysed, I was able to match sizes to content based on said file sizes.
In summary, how I believe Waters Synapt G2-Si data acquisition works is in the following way (I have attached a picture of the experiment setup software in case it helps with visualization):
i. It will create as many functions as different experiments the user selects, with LockSpray quite probably being the last experiment, e.g., if I add two experiments such as full scan MS and full scan MS/MS, with a LockSpray option enabled, it will create function 1 (MS), function 2 (MS/MS) and function 3 (LS).
ii. If I enable ion mobility on the corresponding experiments, then it will create the same 3 functions from point (i), and 3 more: function 4 (IM for MS), function 5 (IM for MS/MS) and function 6 (IM for LS).
Thus, there is nothing set in stone regarding function numbers and their content, but it should be possible to determine in all cases what each function contains by reviewing the _extern.inf file, determining how many experiments were included in sample analysis, and whether ion mobility was enabled or not (which is also included in the _extern.inf file).
I have uploaded the two remaining example files:
- 220505004_HRMS-LS: contains full scan MS and LockSpray data (two functions).
- 220511004_HDMS-LS: contains full scan MS, ion mobility and LockSpray data (four functions).
- 220517008 (previously uploaded): contains full scan MS data (one function).
- 220520007 (previously uploaded): contains full scan MS and ion mobility data (two functions).
Regarding your last question, we are able to open and process all files using Waters proprietary software (MassLynx and DriftScope), so although files could be corrupted, we have no way of evaluating it. I can confirm that all samples within the same sequence analysed with the same experiment setup contain the same number of functions.
Another lengthy response, I apologize if it is too convoluted, do let me know if there is something you would like me to rephrase.
Thank you for looking into this issue. |
|
| |
|
|
 Captura de pantalla 2022-05-25 a las 12.19.32.png
Captura de pantalla 2022-05-25 a las 12.19.32.png Captura de pantalla 2022-05-27 a las 10.02.39.png
Captura de pantalla 2022-05-27 a las 10.02.39.png Captura de Pantalla 2022-06-03 a las 10.19.58.png
Captura de Pantalla 2022-06-03 a las 10.19.58.png Captura de Pantalla 2022-06-03 a las 10.20.43.png
Captura de Pantalla 2022-06-03 a las 10.20.43.png Captura de Pantalla 2022-06-03 a las 10.20.58.png
Captura de Pantalla 2022-06-03 a las 10.20.58.png Captura de Pantalla 2022-06-03 a las 10.21.12.png
Captura de Pantalla 2022-06-03 a las 10.21.12.png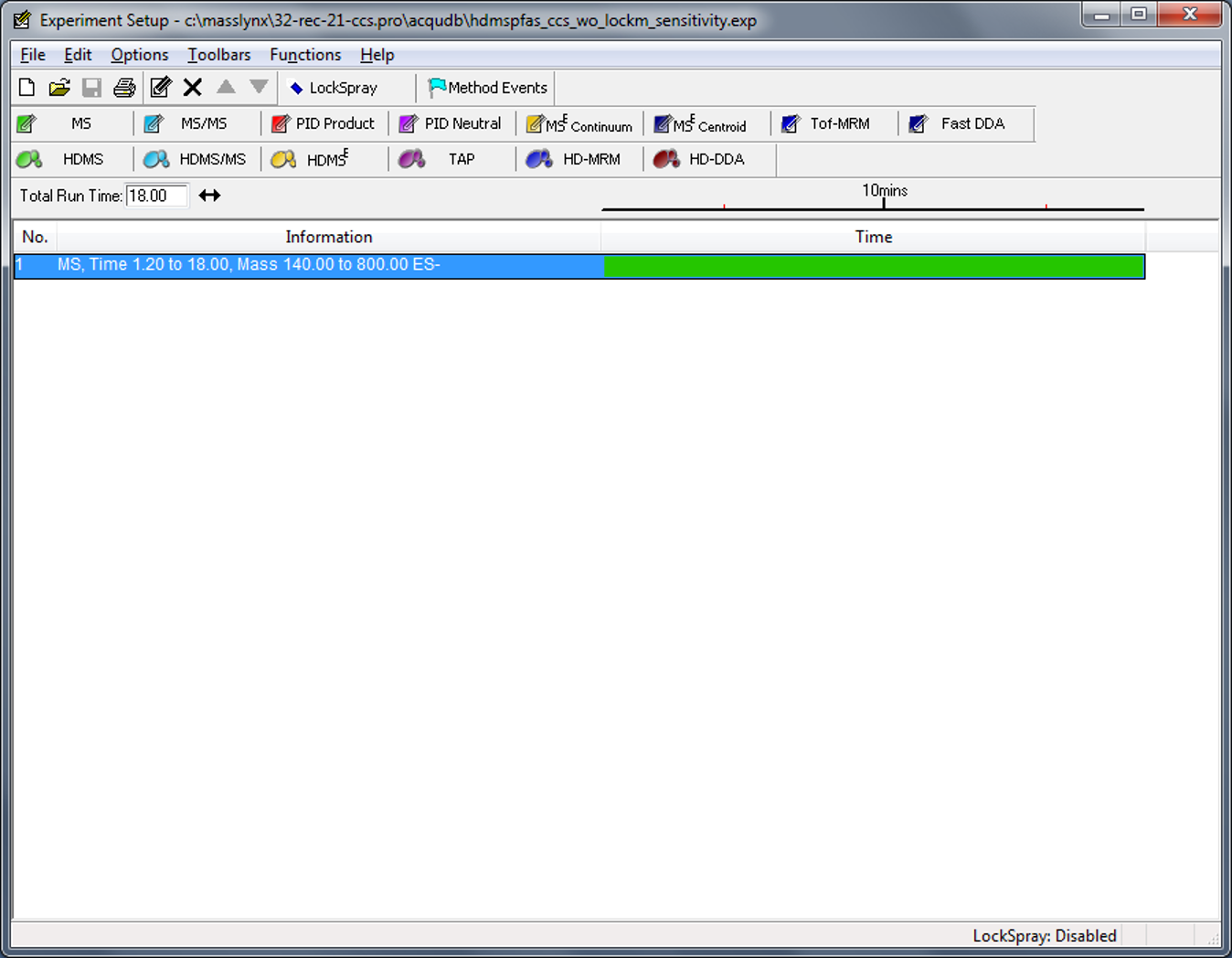 experiment_setup.png
experiment_setup.png