| SWATH Identification | Lehnert | 2019-01-10 06:43 | |||||||||||||||||
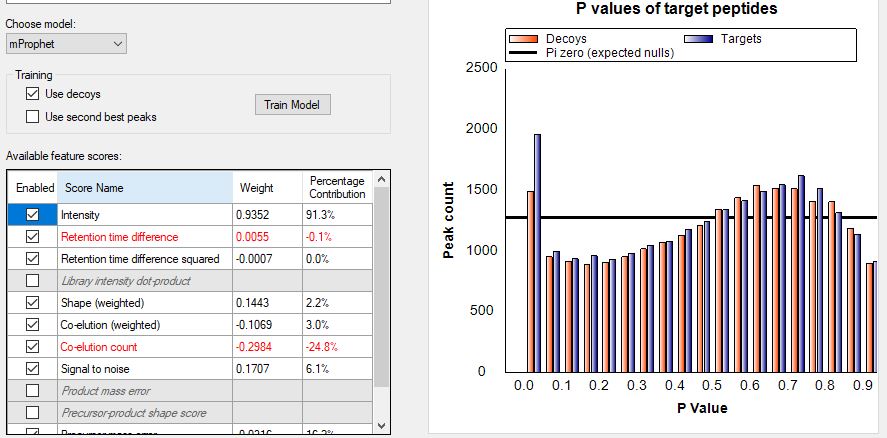
Dear Brendan, we have done a SWATH project with a large library consisting of thousandes of proteins and are now trying to analyze our DIA run (HCP-Project). Unfortunately Skyline identifies and quantifies almost any protein present in the library although for most peptides there is only background noise. So far I am unable to remove those false postitives since I have not found a way to define a peak detection threshold. Is there any way to do that (or what am I doing wrong) and if not what can I do to remove those false positives? Tahnks and best regards, Stefan |
|||||||||||||||||||
| |||||||||||||||||||
 Screenshot (all).JPG
Screenshot (all).JPG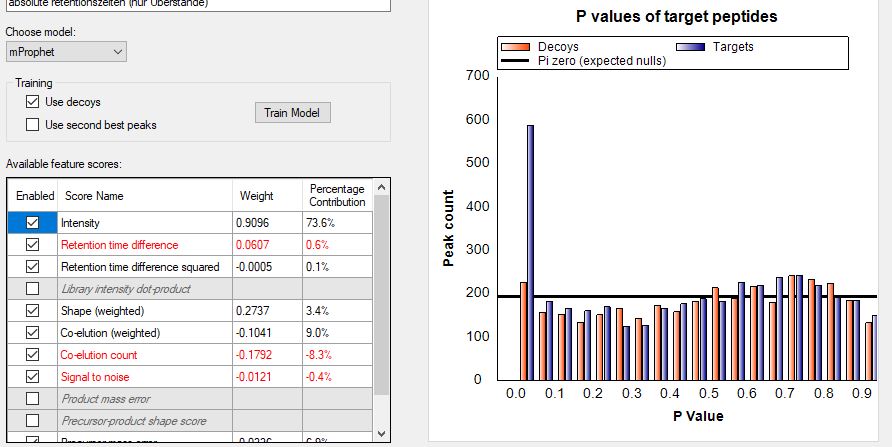 Screenshot (Supernatants only).JPG
Screenshot (Supernatants only).JPG