Hi,
With recent analytical chemistry publication, there's something interesting to do with multiplexed top down PRM (https://pubs.acs.org/doi/abs/10.1021/acs.analchem.8b02699).
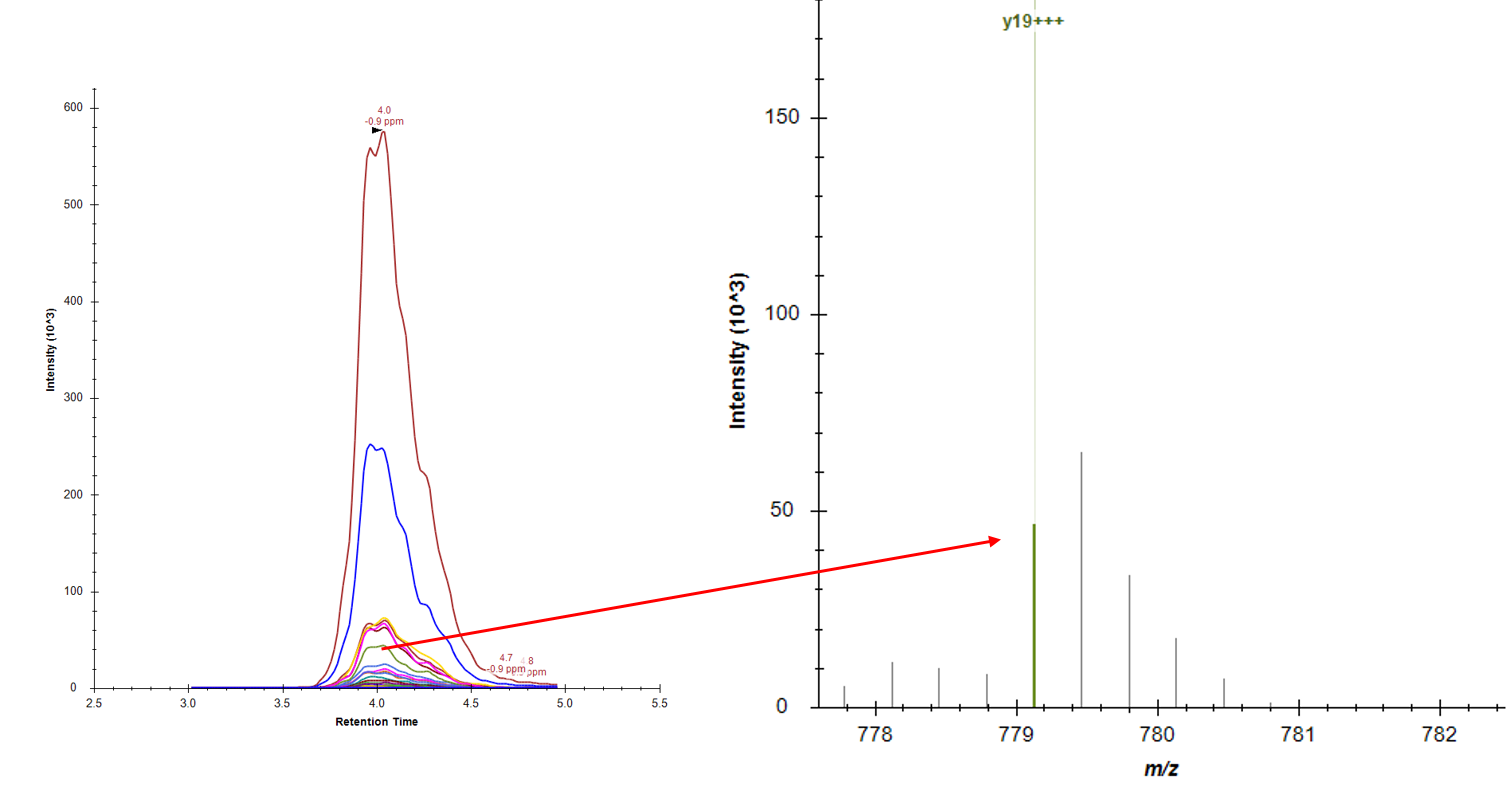
While doing some experiments on my side, I thought this was indeed interesting, but there might be something to add to Skyline to complete data analysis of these experiments. Indeed, in case of high molecular weight (with charge > 1), it's very likely that fragments' most abundant isotope would not be the monoisotopic. However, Skyline seems to integrate only the first one (see capture attached). Maybe this feature is already implemented but I couldn't find it.
By adding all/multiple isotopes to target ions integration, this would increase detected signal intensity which would affect results significantly.
I can send data if needed.
Best regards,
Vivian
|
| |
| Nick Shulman responded: |
2018-10-10 06:03 |
There is currently no way to do this in Skyline.
I actually made a poster about what it would look like if Skyline were extracting the full MS2 fragment isotope envelope from DIA experiments. I was able to find one peptide that looked really good, but for most peptides, I think, looking at more m/z values in the MS2 increased the chance that you will have interference.
With a PRM experiment, the isolation window is usually (I think) .7 units wide. The precursor has to be at least charge 3 in order for anything other than the monoisotopic precursor to have been isolated. The monoisotopic precursor only produces monoisotopic fragments. The M+1 precursor produces a mixture of monoisotopic and M+1 fragments. One interesting feature of extracting the fragment isotope envelope would be for Skyline to report the fragment isotope dot product, but in order to do that, Skyline would need to know exactly how sharp the edges of the precursor isolation window were.
I was not able to find a scenario where this would actually help, but it would be great to take a look at your data.
You can upload your files here:
https://skyline.ms/files.url
If you are sending us your Skyline document, you should use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files, including extracted chromatograms.
-- Nick |
|
| |
| v delcourt responded: |
2018-10-10 06:44 |
Hi,
Thanks for your quick answer. In facts, this is true for peptides, but for large peptides or full length proteins, this feature would I believe be interesting.
Are joined two files with 1 large peptide (> 3 kDa) and one full length protein (21 kDa) with both acquired in multiplexed PRM as shown in publication I mentioned.
Also joined : Xcalibur integration of single isotope of first post mentioned fragment (top) and integration of same fragment but for 5 isotopes (bottom).
Best regards,
Vivian
EDIT : Sorry, I noticed the procedure for creating the zip file after upload |
|
| |
| v delcourt responded: |
2018-11-02 00:41 |
Hi,
Is this feature still in discussion on your side ?
Best regards,
Vivian |
| |
| Martijn van Duijn responded: |
2018-12-11 05:41 |
Hi, I was also trying to integrate top-down PRM data of an intact protein into Skyline. My protein chain of interest is about 23 kDa, multiply charged in ESI. In order to circumvent the isotope problem, I wanted to try to use xtract (xcalibur on orbitrap data) to deconvolute all my MSMS scans of my precursor of interest, e.g. the 16+ of my protein. In the decharged/deisotoped MSMS spectra only a single peak remains for each fragment ion, which should be ideal for skyline. I ran into the problem of the maximum m/z limit that is allowed in Skyline. As detailed in this support ticket Issue 533 the limit is now somewhat arbitrarily set at 10,000. It would be helpful, to me at least, if this could be raised further for that purpose. |
| |
| warham responded: |
2025-05-13 20:01 |
For building explicit transition lists, with known fragment ions, which are large enough and charged enough +3->+9 that the monoisotopic peak is a very small component in the fragment ion isotopic envelope, could you enable ir1-irN calculation for those ions when importing transition lists? |
| |
| Brian Pratt responded: |
2025-05-15 08:04 |
Could you provide an example of what you’d hope to be able to import? |
| |
| warham responded: |
2025-05-16 14:32 |
So I built a transition list (attached) which has a peptide, labeled as a molecule, with explicit precursor charge states and specific product ions associated with the particular charge states. The precursor and the product ions are large and multiply charged. When I import this file into skyline, skyline automatically calculates the isotopic envelope that it expects for the precursor ions, but it imports the product ions as monoisotopic mass only, even though the chemical formula for the product ion is supplied and read in. It would be sooooooooooooo incredibly cool if we could get the isotopic envelope calculated for large product ions also, please, pretty please :) This would make it possible to build transition lists where fragmentation of regioisomers could be predicted and integration of msms could be used to distinguish between regioisomers, robustly, for larger molecules, the way it can for smaller ones, now. and the larger the molecule the more likely it is to have regioisomers, whether it's synthesized, or a peptide with a modification (like oxidation) at different positions. |
|
| |
| Nick Shulman responded: |
2025-05-16 17:37 |
warham,
If you send us your Skyline document and a few of your raw files I could see whether the special version of Skyline that I made in 2018 has any features that would be useful for you.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
The "Share Document" dialog gives you the option of including the raw files in the .zip file or you can send them separately.
Files which are less than 50MB can be attached to these support requests.
You can always upload larger files here:
https://skyline.ms/files.url
-- Nick |
| |
| warham responded: |
2025-05-16 17:45 |
that would be amazing. I think I need the link to a larger file transfer format than the attachments below. |
| |
| Nick Shulman responded: |
2025-05-16 20:15 |
|
| |
|
|
 topdown_prm.PNG
topdown_prm.PNG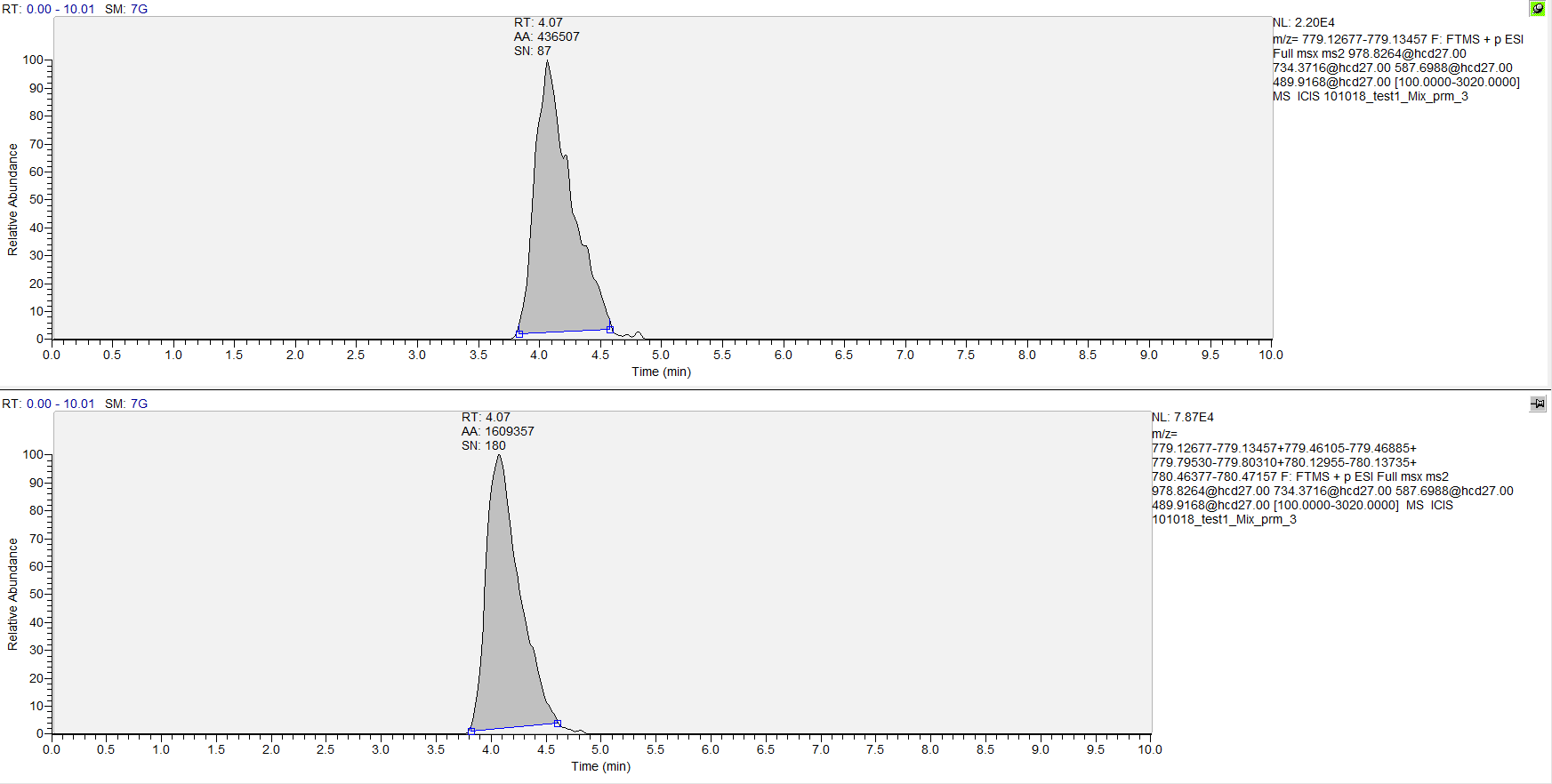 Skyline_topdown_prm2.PNG
Skyline_topdown_prm2.PNG