| Allow integration of multiple isotopes for high molecular weight fragments (Top-Down PRM) | v delcourt | 2018-10-10 03:48 | |||||||||||||||||||||||||||||||
Hi, With recent analytical chemistry publication, there's something interesting to do with multiplexed top down PRM (https://pubs.acs.org/doi/abs/10.1021/acs.analchem.8b02699). While doing some experiments on my side, I thought this was indeed interesting, but there might be something to add to Skyline to complete data analysis of these experiments. Indeed, in case of high molecular weight (with charge > 1), it's very likely that fragments' most abundant isotope would not be the monoisotopic. However, Skyline seems to integrate only the first one (see capture attached). Maybe this feature is already implemented but I couldn't find it. By adding all/multiple isotopes to target ions integration, this would increase detected signal intensity which would affect results significantly. I can send data if needed. |
|||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||
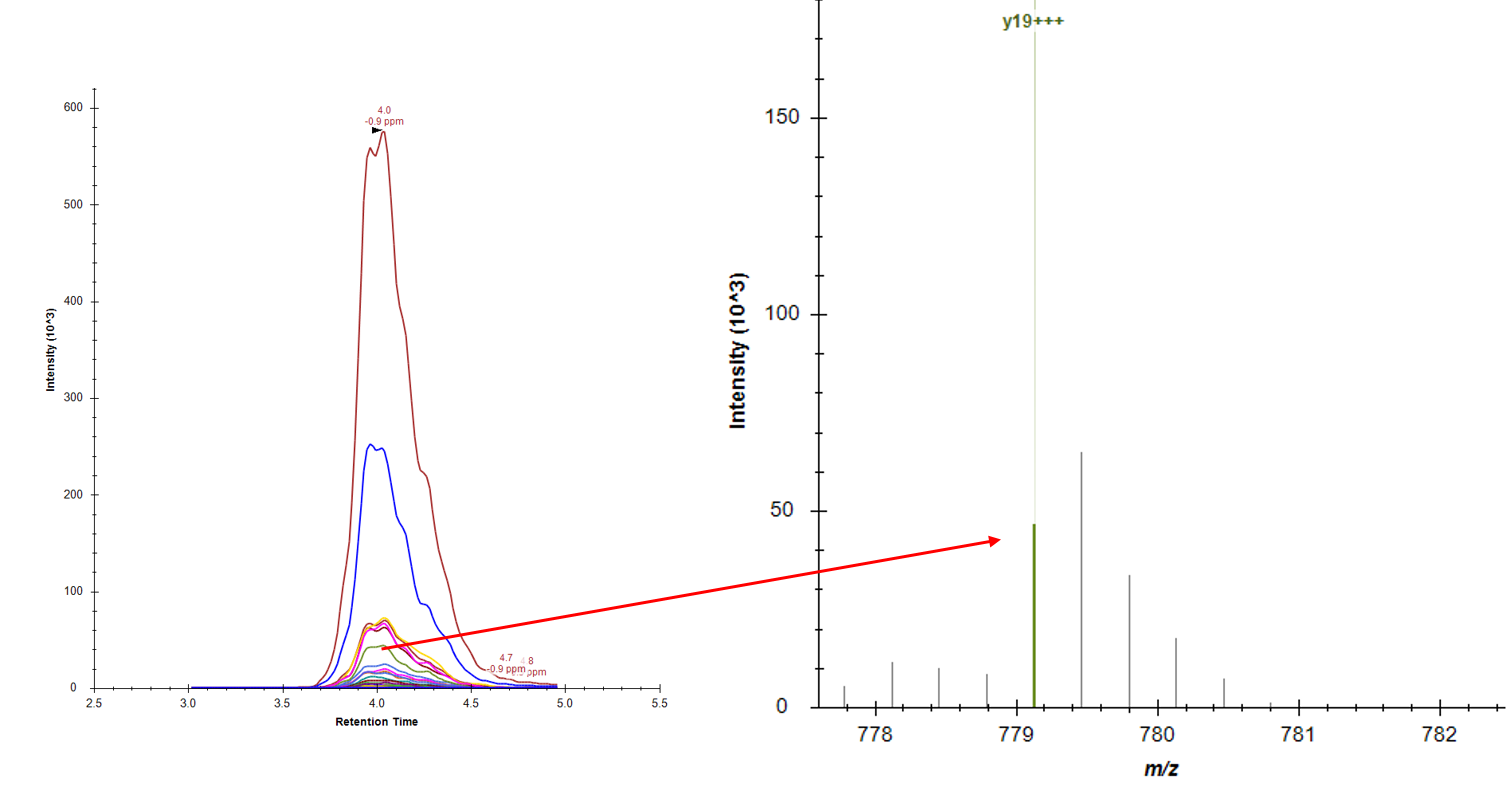 topdown_prm.PNG
topdown_prm.PNG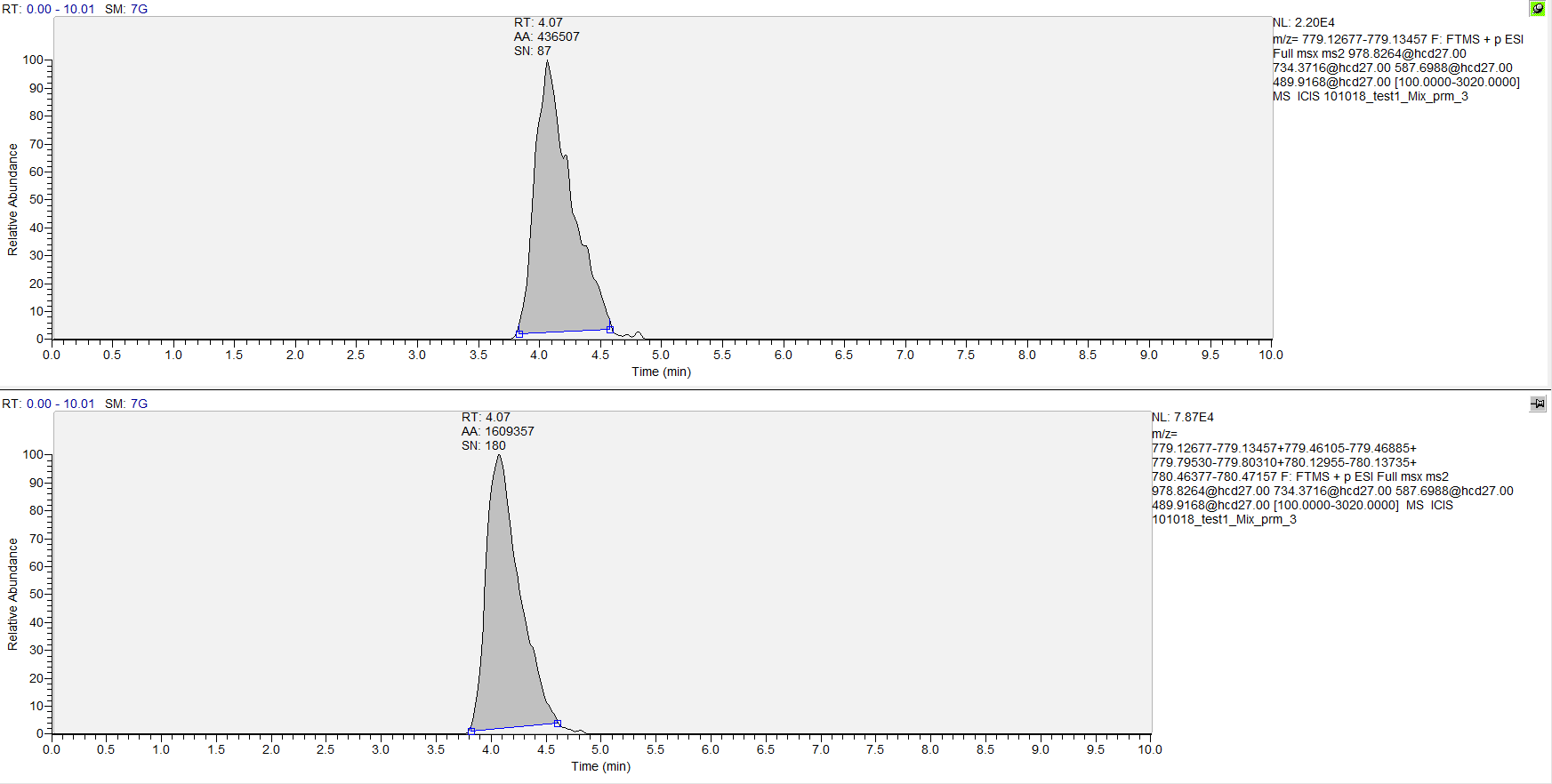 Skyline_topdown_prm2.PNG
Skyline_topdown_prm2.PNG