Sorry if I am too late to this discussion.
When the IM values are extracted from a spectral library does this automatically create an ion mobility library (.imsdb)? Or would this still have to be created through this path:
Settings -> Transition Settings -> Ion Mobility Library -> Add... -> Import... -> "A named spectral library"
Recently I have not been able to use this feature myself too much because the current database search and association software I use don't know how to report IMS. That said, I suspect that if you do it this way there will be cases where a precursor might have multiple IMS values (small decimal differences within the ion's IMS distribution). There will be cases where this fit to multiple gas phase conformers, but I think most instances are the former scenario and I saw that from fragpipe results about a year ago. I think we slightly touched on this on the Dortmund course.
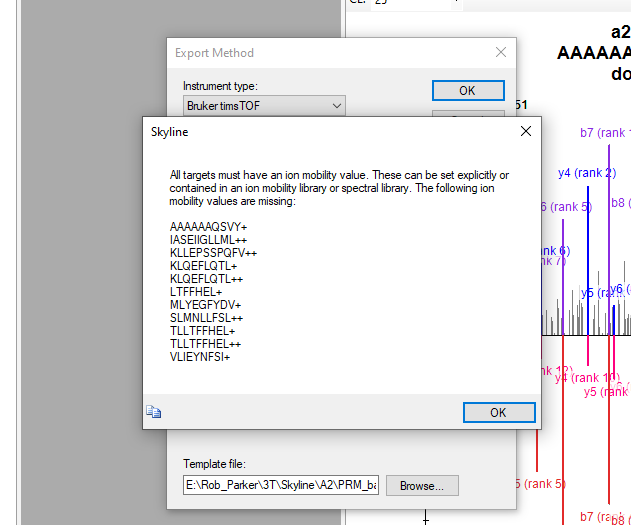
@WeiQiang I have faced the error you had and the way I solve it is by tracking the precursor ions displayed like in the image Robert showed. But you can also create a custom precursor report (see attached figure) to track precursors that are missing IMS values. Perhaps you have some iRT peptides that are not reported in the spectral library?
If you only care for those 12 proteins, for the purpose of generating the method I would delete all other proteins and just keep those you care and make sure the associated precursors have reported ion mobility values.
Hope this helps.
And as always, thank you for your great work Skyline team.
Sincerely,
Juan C.
 Capture.PNG
Capture.PNG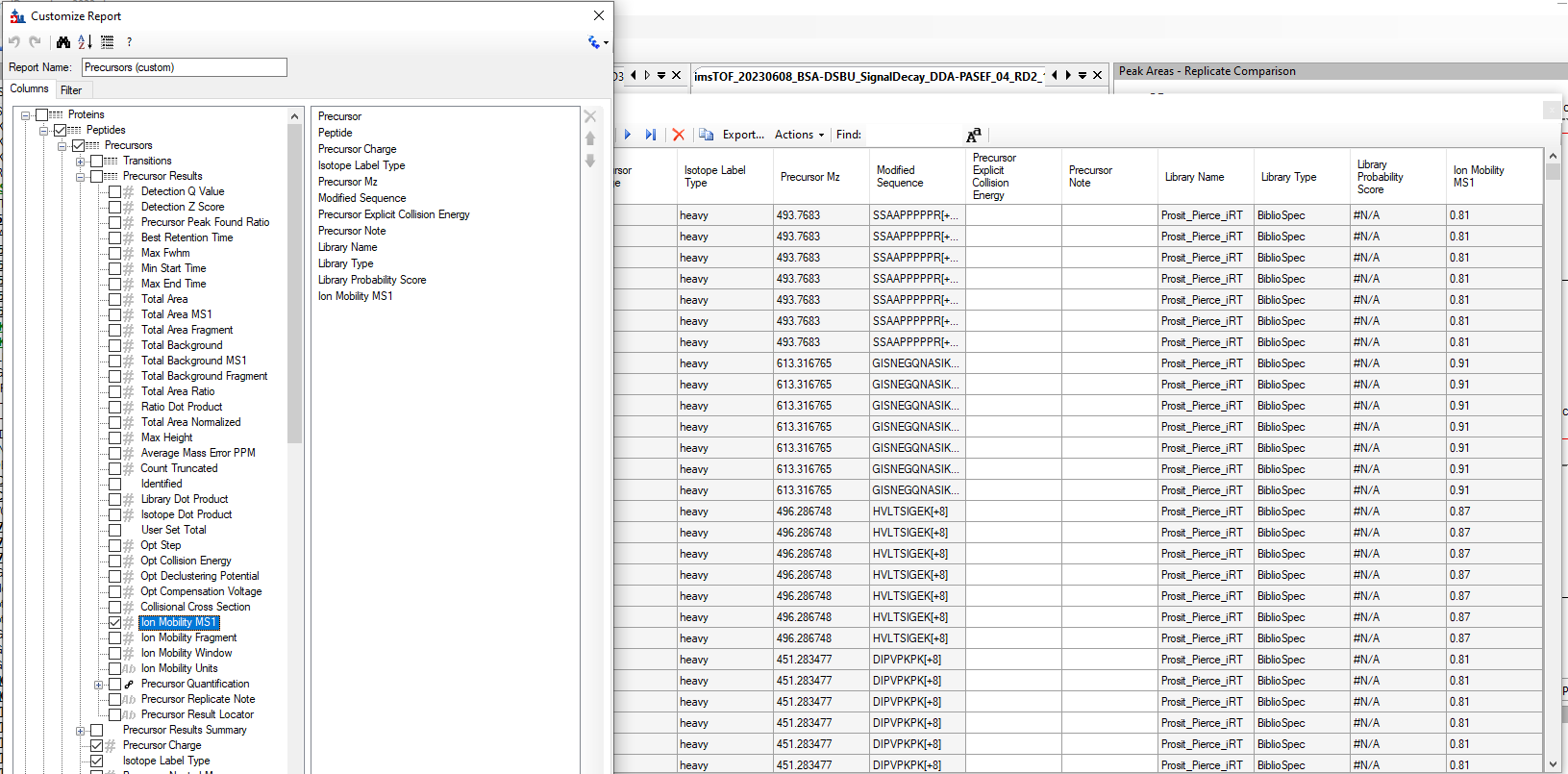 IonMobility_example.PNG
IonMobility_example.PNG