| Nick Shulman responded: |
2022-10-24 12:15 |
Attached is a fixed version of "T_noUV_dim_BPA.sky". You should be able to save it overtop of the file of that name which you already have (so that it ends up in the same folder as "T_noUV_dim_BPA.skyd") and then open it without error in Skyline.
If your peptide search results have obscure modifications, we usually recommend that you go to:
Settings > Peptide Settings > Modifications
and define the structural modification exactly as you would like it to be with the correct chemical formula and amino acids that it applies to.
After you do that, you can do "File > Import > Peptide Search" and Skyline will not need to ask you about the modifications that it finds in the .pep.xml file.
Instead, what it seems happened is that Skyline happened to find a unimod modification which was the right mass.
It sounds like there is a bug in Skyline which allowed a modification to be marked as variable which was not specific to any particular amino acid or terminus. Skyline is not supposed to allow that to happen, but I imagine there's a bug where if the modification gets added in the Import Peptide Search wizard it does the wrong thing.
By the way, if you would like to learn more about modifications in Skyline, in particular explicit and variable modifications, this is a good webinar to look at:
http://skyline.ms/webinar10.url
-- Nick |
|
| |
| lulmer responded: |
2022-10-24 12:45 |
With the updated file, I get the error of "Invalid amino acid 'B.'" I also find that I cannot type B as an amino acid under the Settings > Peptide Settings > Modifications window.
Thanks!
Lindsey |
|
| |
| Nick Shulman responded: |
2022-10-24 12:54 |
Oops. My mistake. Try the attached file now.
-- Nick |
|
| |
| Nick Shulman responded: |
2022-10-24 16:44 |
I think other things are not going to work for you, since you are using "B" as the amino acid, and your peptide search engine's idea of what "B" means is probably different from what Skyline thinks.
Skyline treats "B" as an amino acid whose mass is approximately 114 Daltons, which is almost certainly different from what your peptide search engine was assuming that "B" was.
I think things would work better if you told your peptide search engine that one of the standard amino acids had a variable modification on it whose chemical formula was what it needed to be to change that amino acid into the nonstandard amino acid that you are looking for.
Yes, Skyline does not allow you to specify that a modification applies to "B", "J", "X", or "Z", so it would probably not be possible to get any of those letters to work with your peptide search results.
Let me know if it is not possible for you to apply a modification to one of the standard amino acids instead of using "B".
We might be able to fix Skyline so that it works better with nonstandard amino acids.
I know in the Proforma v2 specification ( https://github.com/HUPO-PSI/ProForma/blob/master/SpecDocument/Release_v2.0/ProForma_v2_Final.pdf ) it says that the amino acid "X" should represent an amino acid of zero mass so that you can easily add a modification to it to represent a nonstandard amino acid. Skyline does not support this, but we could probably implement support for this in a few weeks.
-- Nick |
| |
| lulmer responded: |
2022-10-25 13:50 |
Thanks that file does work for me!
I've found that we get slightly different database search results when we run our searches as a mass shift on an existing amino acid instead of the additional amino acid ''B.' 'B' in our case is the crosslinking reagent benzoylphenelaylanine. There are very different results for crosslinked peptides when running searches with a mass shift on existing amino acids instead of an additional amino acid, so improved skyline compatibility for nonstandard amino acids would definitely be helpful for my use case.
Thanks!
Lindsey |
| |
| Nick Shulman responded: |
2022-10-25 14:32 |
By the way, I am not sure if you are familiar with the crosslinking features in Skyline:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=Crosslinking
Skyline also understands some crosslinking aware peptide search results.
There are some crosslinking peptide search engines listed on this page:
https://proxl-ms.org/
Currently, the only crosslink search results that you can create a BiblioSpec library from are "byonic" and "kojak" (and you have to convert those results to .proxl.xml first) but if you needed support for a different crosslink peptide search engine we could probably add support for it easily.
-- Nick |
| |
| lulmer responded: |
2023-01-09 16:18 |
I've been using the settings from above to view peptides containing "B," and it's been working quite well for non-crosslinked peptides. However, I've had some issues with the crosslinked peptides. I'm using Kojak with Peptide Prophet validation for the crosslink search, and I have been able to successfully convert those results to a proxl.xml, but when I upload the proxl.xml to create a spectral library I get the error "only Byonic or percolator ProxlXML files are supported. Cannot handle search program peptide prophet Kojak” and am unable to create a spectral library. I've also tried manually adding crosslink peptides by using Edit-Insert-Peptides, and the peptides do add successfully, but the added peptides have no children. Any suggestions on how to view these crosslinked peptides would be greatly appreciated.
Thanks!
Lindsey |
| |
| Nick Shulman responded: |
2023-01-09 16:44 |
For each of these sorts of proxl files, we have to do a small amount of coding in order for BiblioSpec to understand them.
Can you send us your kojak .proxl.xml file as well as the mass spec file (.mzML, .raw, or whatever) which was searched?
Those files will probably be to large to attach to this support request so you can zip them up and upload them here:
https://skyline.ms/files.url
-- Nick |
| |
| lulmer responded: |
2023-01-09 17:06 |
Yeah I uploaded the proxl.xml and mzml to that drive as a zip file called Noncanonical_AA_skylinesupport.
Thanks!
Lindsey |
| |
| Nick Shulman responded: |
2023-01-10 08:22 |
Thank you for sending those files.
I see now that BiblioSpec would be able to handle kojak search results, but only if those search results have been processed with Percolator.
Your search results used Peptide Prophet, which we do not yet support in proxl files.
I will fix this.
By the way, I see that in your .proxl.xml file, it says that the mass of your crosslinker is zero:
<linker name="BPA">
<crosslink_masses>
<crosslink_mass mass="0"/>
</crosslink_masses>
That does not look to be correct, and if I were to create a library from that proxl file I don't think it would work right.
Do you think there might be a bug in the kojak to proxl converter that we also need to fix? If so, could you send me more of your files so that we can try to reproduce the problem which is causing that crosslinker mass to be zero?
-- Nick |
| |
| lulmer responded: |
2023-01-10 11:47 |
I intentionally defined the crosslinker mass as 0. The crosslinker I'm using, benzoylphenelaylanine, is a non-canonical amino acid that is incorporated within the protein sequence and has no mass change with the crosslinking reaction. Because of this I've been running the Kojak searches with the mass of benzoylphenelaylanine defined as an additional amino acid "B" with a crosslinker mass of 0. If how I defined this in the Kojak parameters would be helpful, I can send that file.
Thanks!
Lindsey |
| |
| Nick Shulman responded: |
2023-01-10 15:21 |
The next update of Skyline-daily will probably be able to handle PeptideProphet proxl files.
Here is what the .blib file produced from your proxl file is going to look like.
-- Nick |
|
| |
 suggestedmod_01.JPG
suggestedmod_01.JPG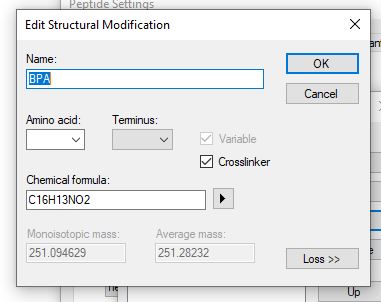 suggestedmod_settings_02.JPG
suggestedmod_settings_02.JPG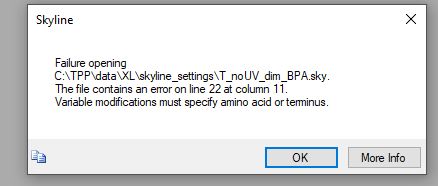 error.JPG
error.JPG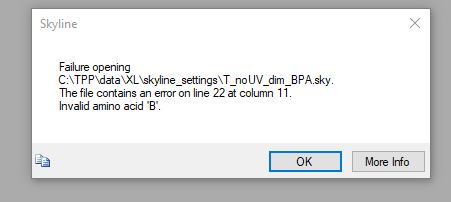 error_02.JPG
error_02.JPG