Can you send us your Skyline document?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
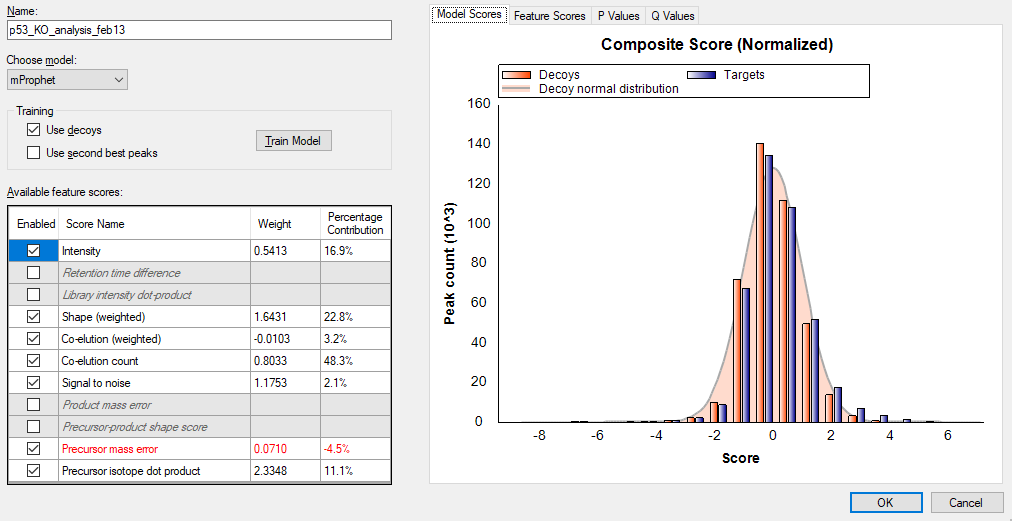
With your mProphet model, the only thing that I could suggest is that you should look into those feature scores which are gray and in italics. Those are features which some but not all of your peptides have and are therefore not available to be used in the peak scoring model. If you select one of those scores, and switch to the "Feature Scores" bar graph, and then hover the mouse below the "Unknown" bar on the graph, you can click on the binocular icon that appears and Skyline will give you the list of peptides that are missing that score. (The list appears in the "Find Results" window which you can get to after you cancel out of the Peak Scoring dialog).
If you remove the peptides from your document that are missing a particular score, that score will become available for model training. That might result in you being able to train a better model.
However, it looks like you already have enough scores available that I would expect your model to look a lot better than it does.
I will probably be able to figure out what is going wrong when I see your .sky.zip file.
-- Nick
 mProphet_model_report.png
mProphet_model_report.png p53_comparison_volcano.png
p53_comparison_volcano.png