| |
| Yao Chen responded: |
2019-08-13 08:01 |
Hello,
I happen to have some experiences with glycopeptide search.
You can go to "Settings -> Peptide Settings -> Modifications" and editing your glycan modifications in the "structrual modifications" window (see picture).
If your glycan modifications are not in the existing list, go to "Edit list->Add->" and you may edit the modifications you want (see picture).
Hope it helps and please let me know if it is still confusing.
Thanks,
Yao
|
|
| |
| Yao Chen responded: |
2019-08-13 08:21 |
After you've edited the modifications, go to "Edit->insert->Peptides..." or "Edit->insert->FASTA..." to define the peptides you are searching.
|
|
| |
| rmeccariello responded: |
2019-08-13 08:34 |
Hi Yao,
Thanks a lot for your response! This sounds like what I do for my SIM files where I'm quantifying based on the parent masses in the MS1. My question is for the analysis of DIA files where I'm quantifying based on the fragment masses (Y1 ion and Y1+Fuc ion) in the MS2. Is there a place I can put in the masses of these fragment ions?
Thanks!
Robin
|
| |
| Brendan MacLean responded: |
2019-08-13 08:40 |
I am not sure I fully understand, but I think I would define 2 structural modifications (Peptide Settings - Modifications) for GLcNAc and make it specific to Terminus = N and one for Fuc and make it specific to Terminus = C. Make both not "Variable" if you really are going to do this for all of your targeted peptides. To the Fuc modification dd a neutral loss of Fuc with "Include loss by default" = Always.
In Transition Settings - Filter, make "Ion charges" = 1, "Ion types" = y, Product ion selection "From" = ion 1, "To" = "1 ion". Make sure all "Special ions" are unchecked.
If you do this, you should be able to add any peptide and have its precursor m/z be "full peptide backbone+full glycan" (where full glycan is GLcNAc+Fuc). Then you would simply need to add your peptides by their sequences and you should get precursors with the desired precursor m/z, containing 2 transitions, one for y1+Fuc (labeled as "y1") and one for y1 (labeled as "y1 -<mass of Fuc>").
Have a look at the attached PowerPoint slides. Even if they don't exactly capture what you had in mind maybe they will help clarify what is possible and give you enough of an idea that you can achieve what you are looking for.
--Brendan
|
|
| |
| rmeccariello responded: |
2019-08-13 09:54 |
Hi Brendan,
Thank you for your response! I think I am half way there. Let me clarify with an example. One of my peptides is EEQYNSTYR. I am looking for this peptide with FA2, or FA2G1, or FA2G2, or FA2G2S1, etc... up to 13 possible N-linked glycans. So one of my parent masses is EEQYNSTYR+FA2. The parent mass +3 charge is 878.68. This mass is in my targeted list, and I have this in Skyline.
In the MS2, the parent mass of 878.68 gives rise to 2 fragment ions that are particularly abundant, so I monitor these. One is the Y1 ion, which is EEQYNSTYR+GlcNAc, and the other is the Y1+Fuc ion (1392, +1), which is EEQYNSTYR+GlcNAc+Fuc (1538, +1). See powerpoint, easier to explain with a picture!
So the masses I want Skyline to monitor for each parent mass are 1392 and 1538. Every glycan, no matter what it is, will give rise to at least one of these two fragments. That's why I want to monitor these fragments in order to quantify the parent masses. But I don't know where I can put in these masses.
I tried to get there backwards by doing as you suggested and inputting the mass that would have to be lost from the glycan in order to result in the breaking of the bond to get the Y1 or the Y1+Fuc.
As it stands, when I try to import a .raw file, I get the error "You must add at least one target transition before importing results".
Any more thoughts would be much appreciated!
|
|
| |
| Brendan MacLean responded: |
2019-08-13 10:13 |
Well, Skyline is right that your targets only contain precursurs and no actual transitions (i.e. the y1 ions you mention are not present).
It seems that you must not have used the Transition Settings - Filter settings that I specified in slide 3 of my original response. However, you will also need to make your modification "Terminus" = C and not "Amino acid" = N. Otherwise, your modification is not included in the y1 ion and therefore cannot be subject to a neutral loss in y1.
Please review the original slides more closely. The omissions you have made are causing at least some of your problems.
Especially, review the final Targets tree in slide 3. You need to end up with both your desired precursor m/z values showing up as the children of your peptides and the desired y1 and y1 -mass values showing up under the precursors.
--Brendan
|
| |
| rmeccariello responded: |
2019-08-14 06:17 |
Hi Brendan,
Thank you for your response. I have made the changes to the modification so that it is on the C terminus. I've attached slides showing the results of doing so. At first I was getting the signal resulting from individual scans and without a scan filter, and then realized that was because I had my acquisition method set incorrectly--I set it to DIA, which is what the Thermo MS software I use calls the experiment, but I should be calling it "Targeted" in Skyline. Doing this got me exactly what I needed.
I also tried getting my Y1 and Y1+F fragment ions monitored by setting them as special ions, and putting my glycans as modifications on "Amino acid" = N, and this got me the same result as far as I can tell. This method seems a bit more straightforward as I do not have to calculate the neutral losses for each individual glycan, or put the modification on the wrong amino acid, but please let me know if I am missing something here and if the first method is superior.
Thank you again so much for your help.
Robin
|
|
| |
| Brendan MacLean responded: |
2019-08-14 08:18 |
Hi Robin,
Looks like you worked out a few issues. I think I misunderstood the meaning of Y1 and Y1+F. I thought by "Y1" you meant the first y-ion in the y-ion fragment series, i.e. Y1+F would be R+F or potentially K+F on a peptide with a C-terminal Lysine. It now seems to me that F1 is a glycan structure and Y1+F is a more complex glycan structure composed of two parts. Sorry, my knowledge of glycans is pretty rudimentary.
So, yes, if you are just trying to measure "charged losses" from the peptide, that is exactly what the Transition Settings - Filter tab Special Ions were created for, and it seems like you have worked out how to use them. I thought you were actually trying to measure something dependent on the amino acid backbone of the peptide (i.e. the y1 fragment + a modification or with that modification absent - a neutral loss).
Your solution looks great, and I will know now not to assume that Y1 and the y1 fragment are the same thing when discussing glycan fragments.
Thanks for your detailed slides and taking the time to explain.
--Brendan
|
| |
| Brendan MacLean responded: |
2019-08-14 08:24 |
Also, the "spiky" pattern of the chromatograms you show in slide 2 is typical of extraction where Skyline is mistakenly trying to extract chromatogram points from spectra that do not measure the target, along with spectra that do actually measure the target. As you found, usually some change to the Transition Settings - Full-Scan tab can fix the problem. Though, we have also seen bugs in our interpretation of spectra among the 6 instrument vendors we support causing this issue. Probably you could have also come up with a DIA isolation scheme that would have worked, but for your purposes, "Targeted" (i.e. PRM) is the right choice.
|
| |
| rmeccariello responded: |
2019-08-14 13:43 |
Hi Brendan,
Ahh! Yes sorry, I should have specified what I meant by Y1 and Y1+F as they are too close to the y1 term. When using HCD, N-glycopeptides can fragment along the glycan more than along the peptide backbone, so what glycoprotein folks call a Y1 ion is the full peptide backbone with the first GlcNAc residue still attached and the rest of the glycan having been fragmented off.
Thanks for verifying the special ions approach. I appreciate your help, as using Skyline for this project is proving to be infinitely more efficient than processing the data via Xcalibur. Thanks for an awesome software and the quick support!
Robin
|
| |
| Brendan MacLean responded: |
2019-08-14 17:43 |
So, does the part that gets cleaved off have a charge? Or is this essentially what is considered a precursor with a neutral loss in phosphoproteomics? You see "precursor -98" a lot in phosphoproteomics.
If so, then you might still be able to do this just by defining neutral losses on your N modification. It seems like maybe there are 2? 1) with the loss of everything but GlcNAc+Fuc, 2) with the loss of everything but GlcNAc?
Since the fragment ion contains the full peptide backbone, approaching things this way would still allow you not to need to define 2 special ions for every peptide you want to monitor.
You are essentially saying 1) the peptide has a glycan modification on N, 2) in CID the glycan modification loses either everything but GlcNAc+Fuc or everything but GlcNAc.
Do I have this right? Or am I still misunderstanding?
Hmmm... Though, looking more closely at your slides again, it looks like you are only monitoring 1 peptide sequence with 3 different Glycan structures on the N residue, in which case Y1 and Y1+Fuc remain constant for all 3 targets, but if you wanted to add, say, 10 more peptides monitored with the same 3 Glycan structures, now you would have to define 20 more special ions, because each special ion contains the peptide backbone. Then it seems like it would be simpler if you had defined the 3 glycan structure modifications as having 2 possible neutral losses.
Or, I guess it does seem like they are charged losses, since you are going from a charge 3 precursor to a charge 1 fragment.
I am glad you now have something that works, but I think if I understand this better, maybe we can make things work even better in the future. It seems like we might want to extend our neutral loss support to charged losses adding a charge loss value in the loss editor, which would obviously be zero for a neutral loss.
--Brendan
|
| |
| rmeccariello responded: |
2019-08-15 06:46 |
Hi Brendan,
Exactly--the peptide has a glycan on N, and the most common fragment to be generated in the ms2 is the full peptide backbone plus a GlcNAc (Y1) or the full peptide backbone plus a GlcNAc and Fucose (Y1+F). The precursor is most abundant as a +3 so I'm monitoring that mass as my parent, and the Y1 and Y1+F fragment ions are most abundant as a +1.
I am monitoring 3 different peptides (actually 2 because one is very low abundance in my current samples). I am looking for 13 glycans that can be present on each of the two peptides. Because the Y1 and Y1+F ions are generated when most of the glycan has been fragmented off, leaving just the common part of the glycan, I can monitor all 13 glycans on the same peptide backbone by just looking at 2 special ions, the Y1 and the Y1+F. When I want to look at those 13 glycans on the other peptide, I need to make 2 more special ions that account for the different peptide backbone.
I think your suggestion of adding charged losses would be great, because then you can actually denote what's happening in the data: the parent mass is detected in the ms1, and when the glycopeptide is fragmented, it loses most of the glycan and leaves behind a full peptide backbone with a common part of the glycan, which is also charged.
I attached a few slides to hopefully show better what I'm talking about. I think the solution of using special ions is great for now, and I look forward to any updates that might come out in the future.
Thanks,
Robin
|
|
| |
| Brendan MacLean responded: |
2019-08-16 17:03 |
Thanks. Again, helpful. Glad you like the idea of supporting charged losses by extending the current support of neutral losses.
Looking at your slides, it looks like the last peptide may be incorrectly defined since it appears to repeat the precursor m/z 4th peptide (922.0411) instead of the pattern of being -10 m/z of the lgG1 version, which would seem to make the expected Q1 start with 976.
|
| |
| Brendan MacLean responded: |
2019-08-17 09:42 |
I am talking with other biopharma researchers who are only interested in monitoring the precursors in MS1. They have opinions about the glycan structures Skyline should support and the naming scheme we should use, but not really anything about fragmentation since they are only interested in MS1.
So, my next question of you: Is this generally applicable that researchers might want to monitor these two "charged loss" precursor ions in MS/MS? Once Skyline supports charged losses would you suggest that all glycan structure modifications we incorporate also include these 2 charged losses (resulting in Y1 and Y1+F)? If so, what is the chemical formula for the resulting atoms added to (or subtracted from) the peptide backbone for Y1 and Y1+F? If I know what the end result addition/subtraction is, then I can work out the lost atoms for each glycan.
If, however, you think different glycans will yield different best fragments to monitor in MS/MS, do you have any insight into how we should determine potential losses to add to the glycan modifications that might be added to Skyline? I guess you mention Glyco Workbench and Oxford nomenclature. Care to point to any reference list of these structures you feel would be ideal to have easily available in Skyline?
Thanks for all your feedback.
|
| |
|
|
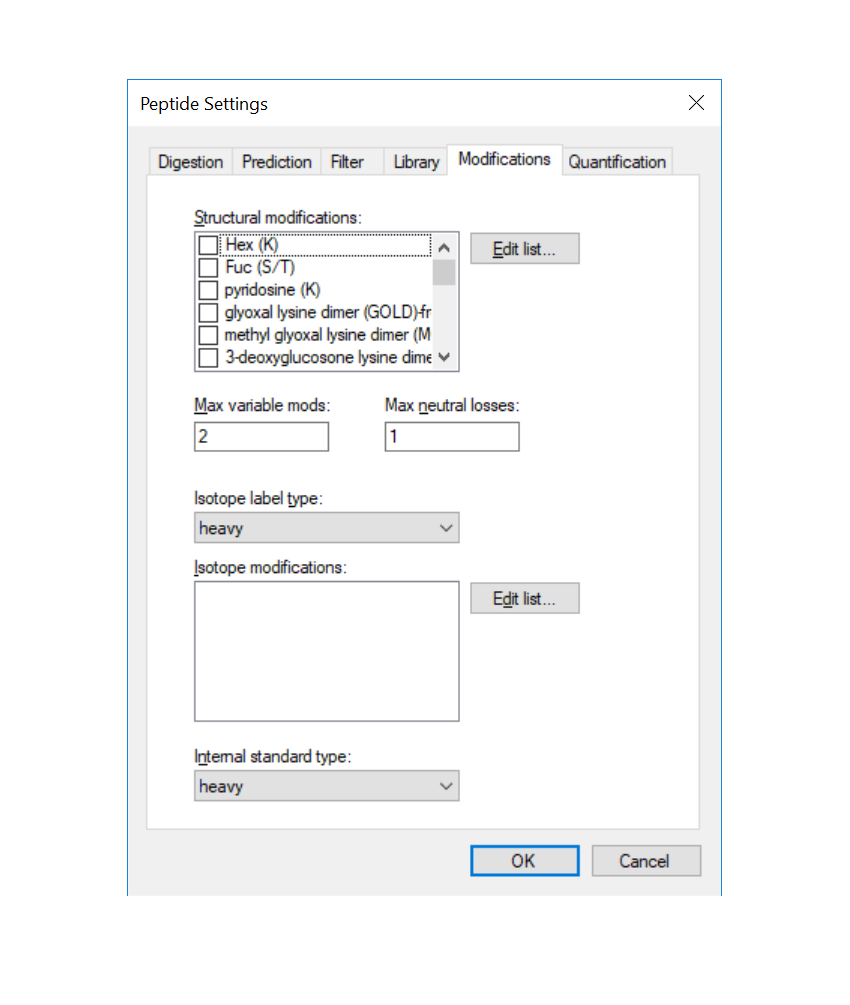 structural modifcations.PNG
structural modifcations.PNG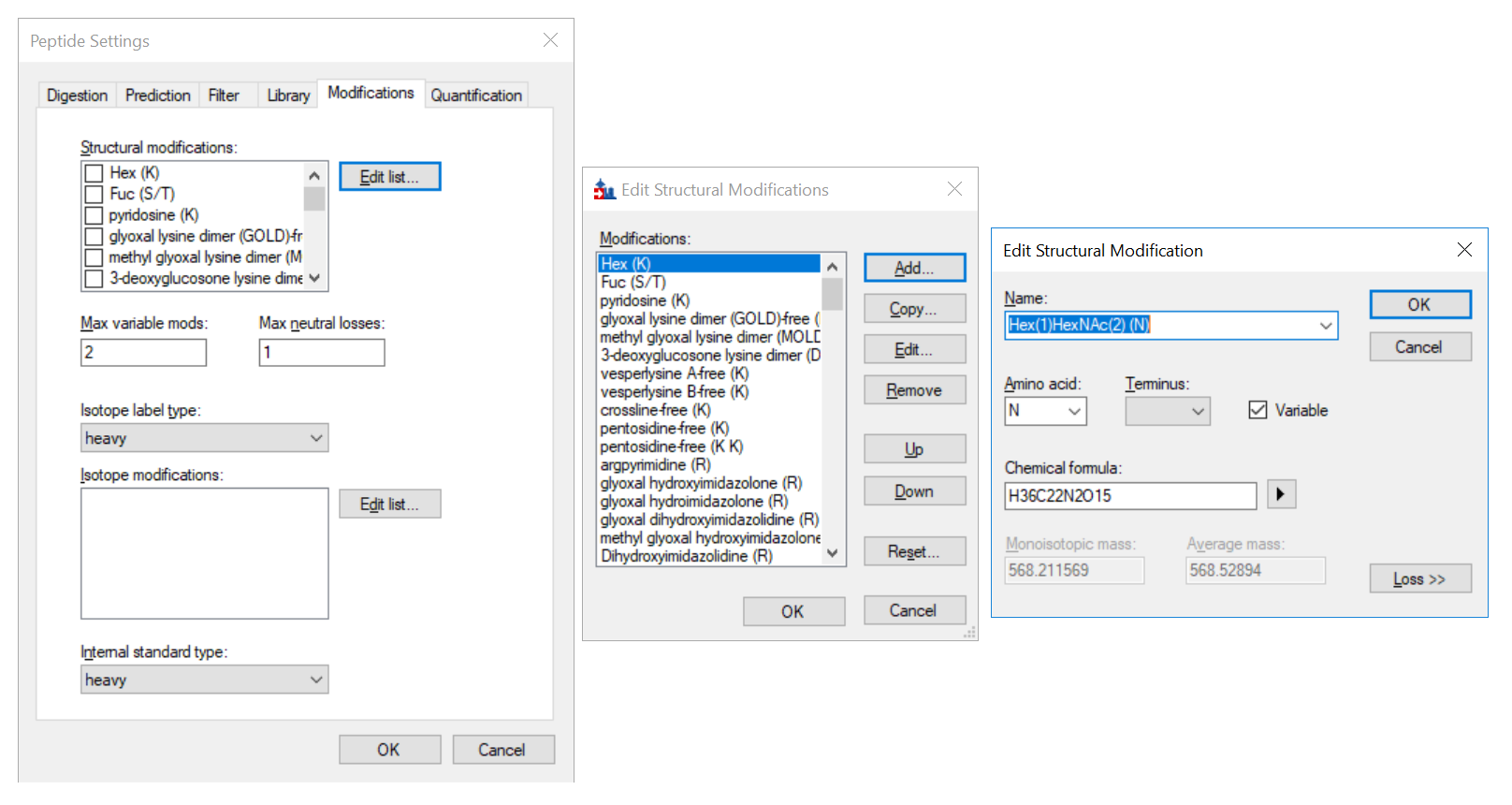 edit structral modification list.PNG
edit structral modification list.PNG insert peptide.png
insert peptide.png