Dear all,
I am using Skyline to Analyze a PRM run (1 MS1 followed by 10 PRM events) of a simple heavy spike-in peptide kit. The peptides have a heavy K8 or R10 at the C-terminus. My example peptide is HVLTSIGEK* with a heavy K8. I imported the peptide search from MaxQuant (1.5.1.0).
Peptide settings:
- Filter: 7 to 25 AA, Exclude N-terminal AA 0, No peptides excluded, Auto-select all matching peptides is ticked
- Library: Generated from the same MQ search, Pick peptides matching: Library, Rank by intensity
- Modifications: No structural mods, 3 varaible mods, 1 max neutral loss, Isotope label type "heavy", ISotope modifications Label:13C(6)15N(2) (C-term K), Label:13C(6)15N(4) (C-term R), Internal standard type "heavy".
Transition settings:
- Filter: Precursor charges 2,3; Ion charges 1,2; Ion types y,b,p. From ion 1 to last ion, no special ions selected, precursor exclusion 5, Auto-select all matching peptides is ticked
- Library: Ion match tolerance 0.5 m/z, If a library spectrum...is ticked, Pick 5 product ions, From filtered nion charges and types plus filtered product ions
- Instrument: 50-1500 m/z, Method match tolerance 0.05 m/z (or 0.1 m/z)
- Full-scan: appropriate settings for the PRM method selected
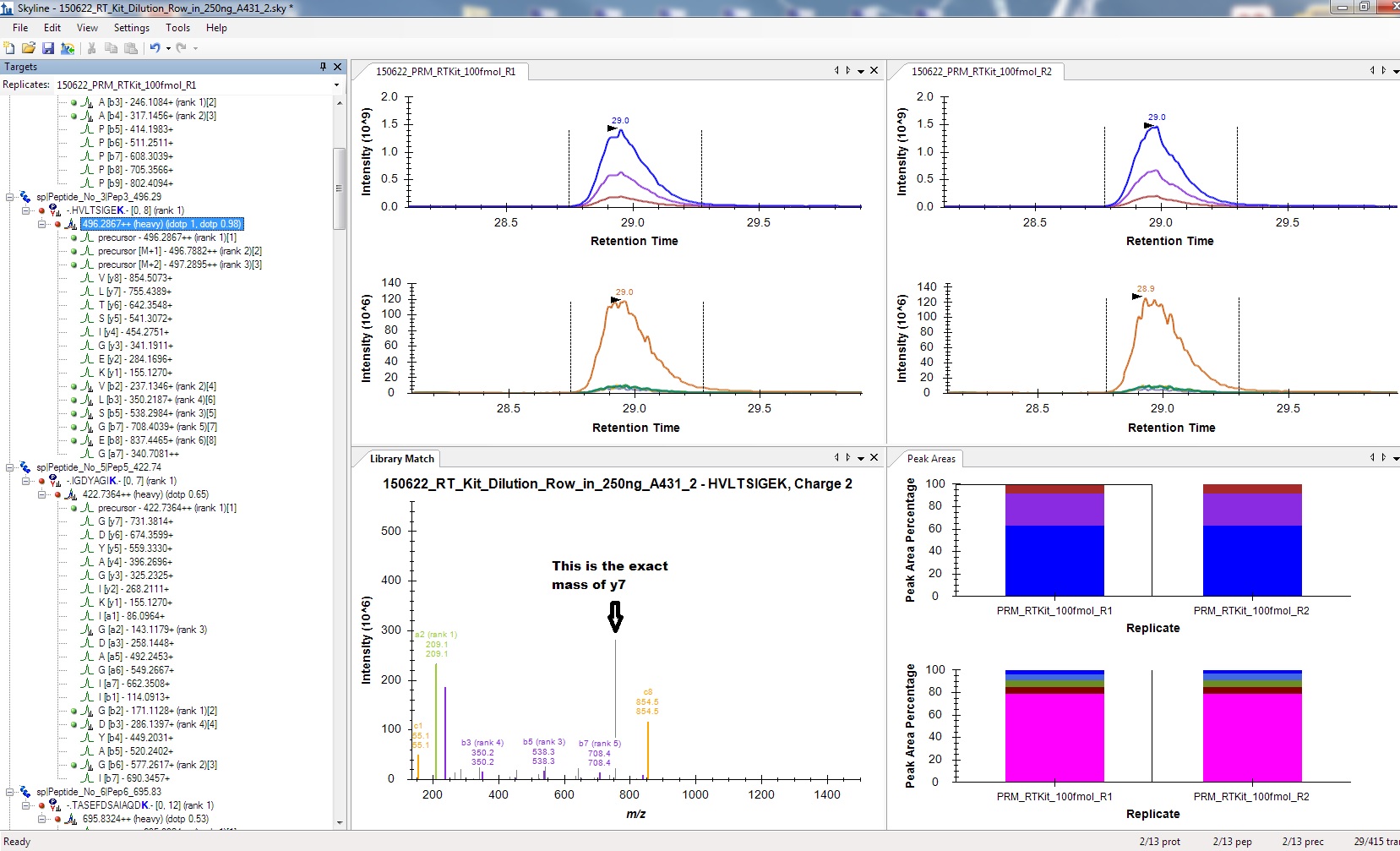
My problem is, that Skyline only utilizes the b-ions (or a-ions). Y-ions do exist in the spectra and are recognized by MQ. Skyline does also understand that the y-ion series will contain the heavy Lysine at the C-terminus and calculates the correct expected y-ion masses. These masses are identical to the MQ msms.txt. Still, Skyline does not allow me to use any y-ion from any of the peptides (see attachment for an example).
Did I choose a setting wrong?
I am grateful for input on this cause
Thanks
Daniel |
| |
| Brendan MacLean responded: |
2015-07-03 05:04 |
Hi Daniel,
I can only think that you added the y-ions after you imported the data initially. If you add ions in Skyline, you need to re-import the data to get Skyline to extract chromatograms for them. In the case you are showing, Skyline would always extract something for all ions included in your method. If there were no signal it would show a red dot, and not just blank space, as you are showing in your screenshot. Blank space means that Skyline has not extracted a chromatogram at all for the transition.
You should be able to use Edit > Manage Results - Re-import to have Skyline perform a new round of chromatogram extraction, including the y-ions in your document. Or certainly, if you remove all results and re-import them from File > Import > Results, that should also get you chromatograms for all of the transitions any precursor where you get chromatograms extracted.
Thanks for posting your question to the Skyline support board. I hope this information gets you past your current issue.
--Brendan |
| |
| danielz responded: |
2015-07-17 01:14 |
Hi Brendan,
thank you very much for your answer. Re-import indeed fixed the problem...but only if I do not specify a spectral library. The problem seems to be that Skyline does not realize that I'm using a spike-in kit that contains C-terminal heavy R10 or K8. When importing the library, Skyline only recognizes b and a ions, not a single y ion (since all should be be 8 or 10 Da heavier) is matched. Skyline calculates the correct heavy y-ion mass and the peak is clearly visible in the spectrum. Still, only b and a ions are used for quantification etc, making data analysis difficult right now. The settings are the same as reported above, y-ions are allowed as Ion-Types.
I'm searching my PRM runs with MaxQuant (1.4.0.5 as well as the current 1.5.2.8) with K8 and R10 as labels and import the msms.txt for the library.
Everything works out fine, if I use non labeled peptides...so I guess the problem is either the MaxQuant output or somewhere in my settings.
Any more ideas what might be wrong?
Thanks a lot
Dan |
| |
| danielz responded: |
2015-08-12 02:12 |
Hi Brendan,
FYI this is a MaxQuant specific problem, when generating the library's with Mascot everything works fine. So i'll switch to Mascot for now.
Best Daniel |
| |
| fredrik edfors responded: |
2015-08-26 08:03 |
Hi Brendan and Daniel,
I'm currently experiencing the same problem as Daniel with loss of y-ions when building spectral libraries from MaxQuant using the msms.txt files. Is this something that you're working on or is this something that the MaxQuant developers have to work on?
Tanks for a great software. Cheers,
Fredrik |
| |
| Kaipo Tamura responded: |
2015-08-26 13:49 |
Hi Fredrik,
To help narrow down the problem, would you mind verifying that the y-ions appear in the "Masses" and "Intensities" columns of the msms.txt file?
Thanks,
Kaipo |
| |
| fredrik edfors responded: |
2015-08-27 00:59 |
Hi,
There are in several y-ions in these columns with reported intensiteis if you inspect the msms.txt file manually. However, only the b-ions are extracted correctly and highlighted in purple in the Library Match window in Skyline. The y-ions are also imported, but they are displayed as grey peaks in the Library Match window.
I've attached one of my msms-files as an example.
Best,
Fredrik |
|
| |
| mscholz responded: |
2016-03-04 00:41 |
Hi,
I stumbled upon this thread when I faced the same problem like Daniel and Fredrik. It turned out that I forgot to set multiplicity to 2 in MaxQuant. If you leave it at 1, MaxQuant assumes you are doing label-free quantification even if you have set a label. As a result the labelling state is not included in msms.txt and Skyline thinks that all identifications stem from non-labelled peptides.
Cheers,
Martin |
| |
|
|
 y-ions.jpg
y-ions.jpg