Hello there!
Would it be possible to define an additional attribute for modifications to define them as different isomers? This is applicable for glycoproteomics, but it should also be applicable to other modification types based on some of the talks I saw in Skyline Online 2025.
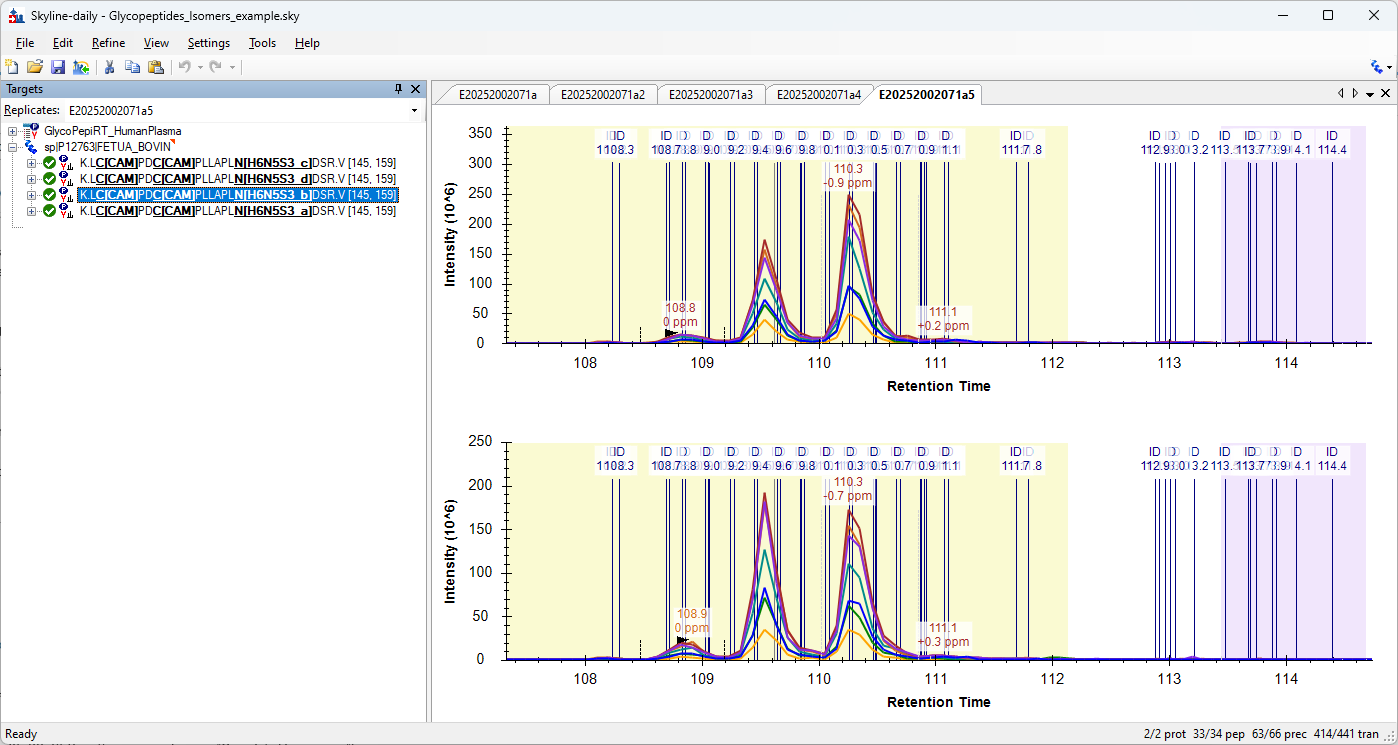
In glycoproteomics, you commonly see glycopeptide structural isomers due to different arrangements of the connectivity between glycan monomers. We can resolve these chromatographically in many instances and would like to quantify them independently.
We have found a workaround for this by creating multiple entries of modifications where the only different thing is the name of the modification. Example:
H6N5S3_a
H6N5S3_b
H6N5S3_c
Here the letter suffix just indicates the elution order. This allows us to create different targets separated by the "Peptide Modified Sequence Three Letter Codes" column in the Document Grid reports.
However, we are having issues with Skyline recognizing these modification isomers when it comes to re-importing peak boundaries and defining independent entries in an iRT library since modified peptides are written with respect to modification mass.
For example for the peak boundaries case, all boundaries of the isomers are fixed to the boundaries of only one isomer after importing a "Peak Boundaries" report.
I am not sure what is the best support for this in Skyline, but we thought maybe an additional attribute written in the Skyline XML files and displayed in the GUI indicating "Isomer = TRUE/FALSE" + choosing which (e.g. user defined as "a", "b", "c", etc.) could work?
We are happy to provide some example data if it would help.
Looking forward to hearing back from you.
Sincerely,
Juan C.
 GlycopeptidesIsomersExample.png
GlycopeptidesIsomersExample.png