| |
| Nick Shulman (Claude) responded: |
2026-01-12 09:17 |
With PRM data, a truncated peaks happen when the time range over which the mass spectrometer was acquiring data for the specified precursor does not entirely cover the time range over which the analyte was eluting. That is, the chosen peak is either at the beginning or the end of the extracted ion chromatogram.
The usual way to fix this is to use wider retention time windows in the acquisition method and run your experiment again.
If you send us your data files we might be able to give you better advice.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
The Share Document dialog gives you the option to include the raw files in the .zip file. Alternatively, you could send those .raw files to us separately.
You can upload your files here:
https://skyline.ms/files.url
-- Nick |
| |
| hadi pourhadi responded: |
2026-01-12 09:42 |
Hi Nick
I hope you are doing well!
I uploaded "2026012_TruncatedPeak_HP"
If it is like that, shouldn't I observe it in all peaks? Please check the pyr-Apelin-13 (511.9418) and pyr-Apelin-13MonoOx(M) 388.2069
Best,
Hadi |
| |
| Nick Shulman responded: |
2026-01-12 09:59 |
Thank you for uploading your data.
You should go to the "Instrument" tab at "Settings > Transition Settings" and uncheck the "Triggered chromatogram acquisition" checkbox.
After you do that you should go to "Edit > Manage Results" and use the "Reimport" button to tell Skyline to extract chromatograms again.
That "Triggered chromatogram acquisition" checkbox should only be used if you are doing a SureQuant method where you have told the mass spectrometer to start collecting data for some precursors when it detects a specific signal such as the heavy precursor. When that checkbox is checked, Skyline will try to guess the times over which the mass spectrometer was collecting data for your precursor. Sometimes Skyline guesses wrong, which results in a chromatogram with gaps in it.
-- Nick |
| |
| hadi pourhadi responded: |
2026-01-12 10:04 |
Thank you for your reply.
In PRM method, I put the masses for precursors for example the 511.9418 and 384.2081 for the Pyr-Apelin -13 and the 517.2734 and 388.2069 for the oxo form of it.
Do I still need to uncheck Triggered chromatogram acquisition?
Best,
Hadi |
| |
| Nick Shulman responded: |
2026-01-12 10:30 |
In a SureQuant method you typically tell the mass spectrometer to start collecting MS2 spectra for the light precursor when it first notices MS1 signal for the heavy precursor. That's not what you did in this experiment, so you should uncheck that "Triggered chromatogram acquisition" checkbox.
In an ordinary PRM method, you tell the mass spectrometer the actual start and end retention times for each precursor, and each precursor has only one start and end time.
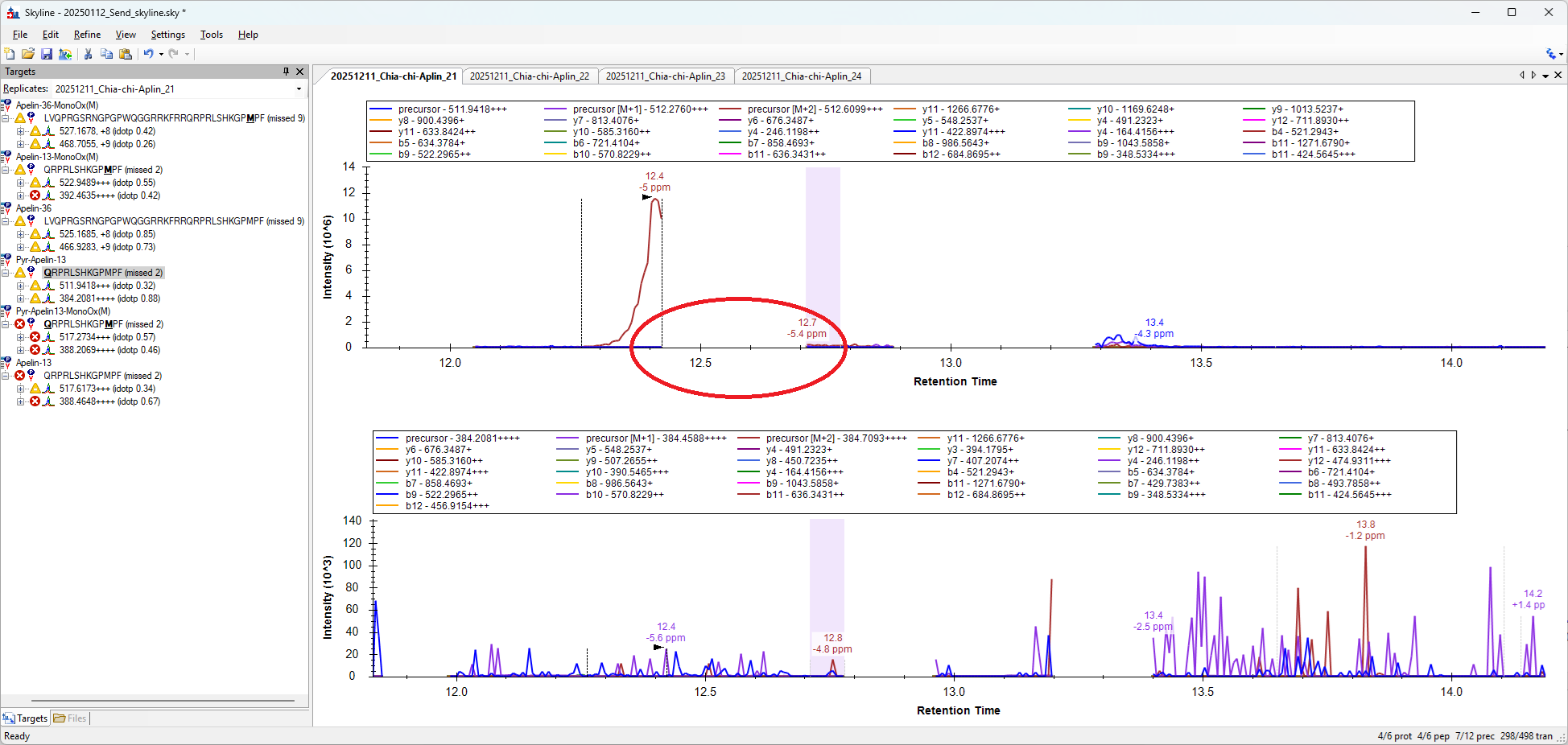
That said, the gaps that Skyline is putting in these chromatograms do represent relatively long time ranges over which no MS2 data was collected for a particular precursor. In the attached screenshot, it does look like the 20 second period from 12.4 minutes to 12.7 minutes there were no MS2 spectra for the 511.9418 precursor. Sometimes gaps like that indicate that it was a DDA method and not a PRM method.
Can you send me your raw file "20251211_Chia-chi-Aplin_21.raw"?
-- Nick |
|
| |
| hadi pourhadi responded: |
2026-01-12 11:14 |
The size of it around 1.3 GB |
| |
| Nick Shulman responded: |
2026-01-12 12:44 |
Thank you for uploading that .raw file.
Can you tell me more about your acquisition method?
The precursor m/z's of the MS2 spectra are almost never identical to each other, such as 384.20233 and 384.20282. This is often indicative of it being DDA data, except that the mass spectrometer does not choose to fragment what I see are the largest peaks in the MS1 spectra, so it was either told to exclude those particular m/z's, or it was somehow told to focus on your m/z's of interest.
We do also see that sort of inconsistent isolation window target m/z values with SureQuant methods, because the mass spectrometer records the m/z that it saw that triggered the MS2 spectrum acquisition, rather that the exact m/z that it was told to look for.
Ideally, when working with PRM methods in Skyline, the isolated m/z's of the MS2 spectra would all be identical when they are targeting the same molecule.
You might need to make the "Method match tolerance m/z" a larger number. That setting is also on the Instrument tab at "Settings > Transition Settings". You have it set to "0.007" (it probably got set that way when you chose "SureQuant" as the "MS/MS Acquisition Method" on the "Full Scan" tab). The default value for that setting is 0.055.
-- Nick |
| |
| hadi pourhadi responded: |
2026-01-14 11:17 |
Hi Nick,
thank you for your respond.
Please find the attached document. |
|
| |
| Nick Shulman responded: |
2026-01-14 11:35 |
Thanks for attaching that file.
I believe that this is a DDA method with an inclusion list, which is not ideal for producing spectra that Skyline can extract chromatograms from.
In a real PRM method, the mass spectrometer would be told to isolate the same precursor m/z's regardless of what is observed in the MS1 spectra. This is what you want to do because some of the things that you want to measure with PRM cannot be seen in MS1 because there is too much signal from other molecules and the mass spectrometer cannot see your precursor of interest among all that noise. The other reason is that you want the spectra that the chromatogram is extracted from to be evenly spaced, or at least not have huge gaps in it so that when you calculate the area under the curve you get the right number.
In this DDA method with an inclusion list, the mass spectrometer was told to look for the m/z's in the list, but only fragment the m/z's that it sees. This is not ideal because the m/z's that get recorded in the raw file end up being the m/z that was seen in the MS1 spectrum, rather than the m/z that the mass spectrometer was told to look for. This means that you have to set the Method Match Tolerance M/Z transition instrument setting to be a high enough number to encompass the difference between the m/z in the inclusion list and the m/z that was observed. But, the bigger problem is that the mass spectrometer will not be isolating particular precursors on a reliable schedule.
I don't know how to use a mass spectrometer, so I can't say exactly what you need to change to turn this into a real PRM method, but I hope it won't be too difficult to figure out.
-- Nick |
| |
|
|
 ChromatogramWithGaps.png
ChromatogramWithGaps.png