| Calibration Curve LLOD and LLOQ | sam lord | 2025-06-04 01:46 | |||||||||||||||||||||||||||||||||||||||||
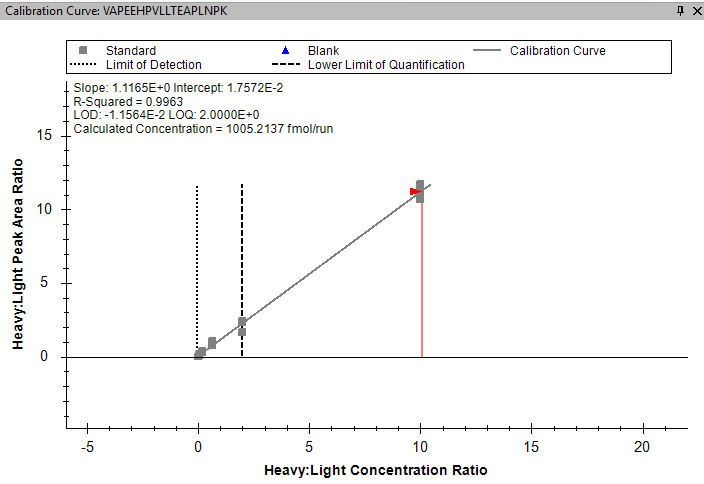
Hello Skyline, I am trying to calculate the LLOQ and LLOD for my heavy internal standards from my calibration curve. I am using a set concentration of light internal standard to normalise the peak area of the heavy peptide. Therefore in the peptide settings in skyline I have set "Internal Standard Type" to Light and the "Normalisation Method" to Ratio to Light. When I look at the standard curve it correctly shows the peak area (y-axis) as a Heavy to Light Ratio, but it also shows the concentration (x-axis) as a Heavy to Light Ratio - See image attached. Ideally I don't want the concentration to be normalised against the Light as I want to work out the LLOD and LLOQ of the heavy concentration. Is there a way to change this so the concentration axis is just showing the Heavy? Many thanks, Sam |
|||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||
 CalCurve.png
CalCurve.png