| Peak Integration: PRM | owagne2 | 2025-04-29 13:42 | |||||||||||||||||||||||||||
Hello, So, my question is this: How do I handle this peak integration, if at all? From where I'm sitting, the endogenous peak doesn't appear to be quantitative, but I'm not familiar enough with Skyline to figure out if the problem is a lack of knowledge on my end, a sample degradation problem, a meaningful biological artifact, or a methods/sample preparation problem. For reference, I am running a PRM proteomics experiment on Skyline version 24.1.0.414. I've attached images of the heavy peak, the endogenous peak that Skyline recommended I integrate, the endogenous peak that I manually wondered if I should integrate, and an overlap of the precursor ions for both the heavy and light peptides (blue is the heavy, red is the endogenous). I first assumed that I have a PTM on my endogenous peptide, but that would change the overall mass and my PRM scan wouldn't have picked up my peak. I have an amino acid in my sequence that perhaps could have undergone racemization (my cell pellets were >1 yr old), but I wasn't sure if there was a simpler or more likely explanation before I attempt a new experiment. For reference, this peptide comes from a membrane bound glycoprotein. To my knowledge, there are no known glycosylation or phosphorylation sites on my peptide. I apologize for the novel, but I thank you for your time and look forward to your response. Sincerely, |
|||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
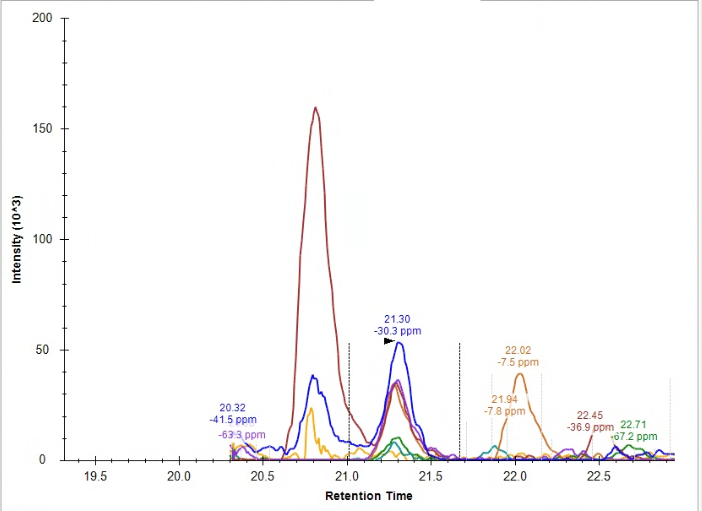 Endogenous_SkylinesPeakChoice.png
Endogenous_SkylinesPeakChoice.png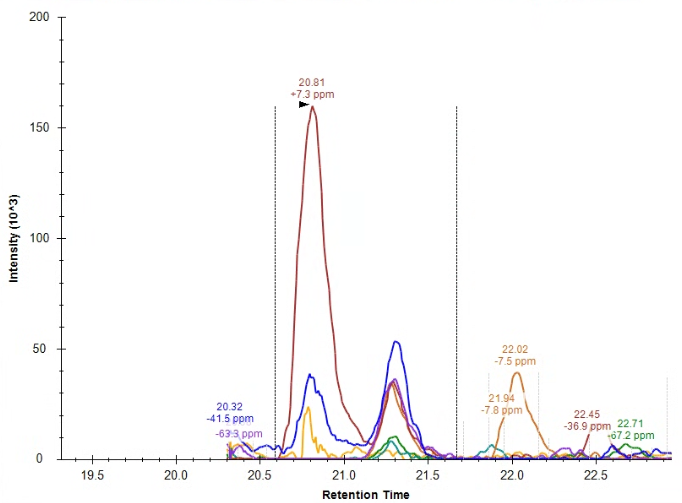 Endogenous_ManualPeakChoice.png
Endogenous_ManualPeakChoice.png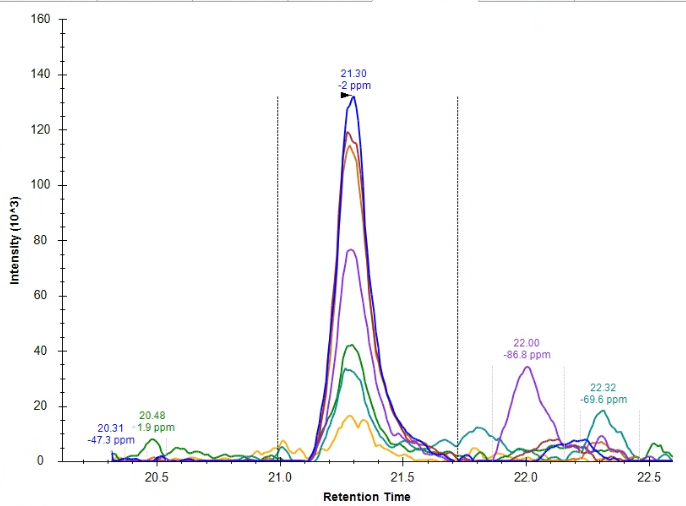 Heavy.png
Heavy.png Overlap_BlueHeavy_RedEndogenous.png
Overlap_BlueHeavy_RedEndogenous.png