| Skyline for small molecules : MS/MS visualisation | oceane delos | 2025-03-25 08:28 | |||||||||||||||||||||||||
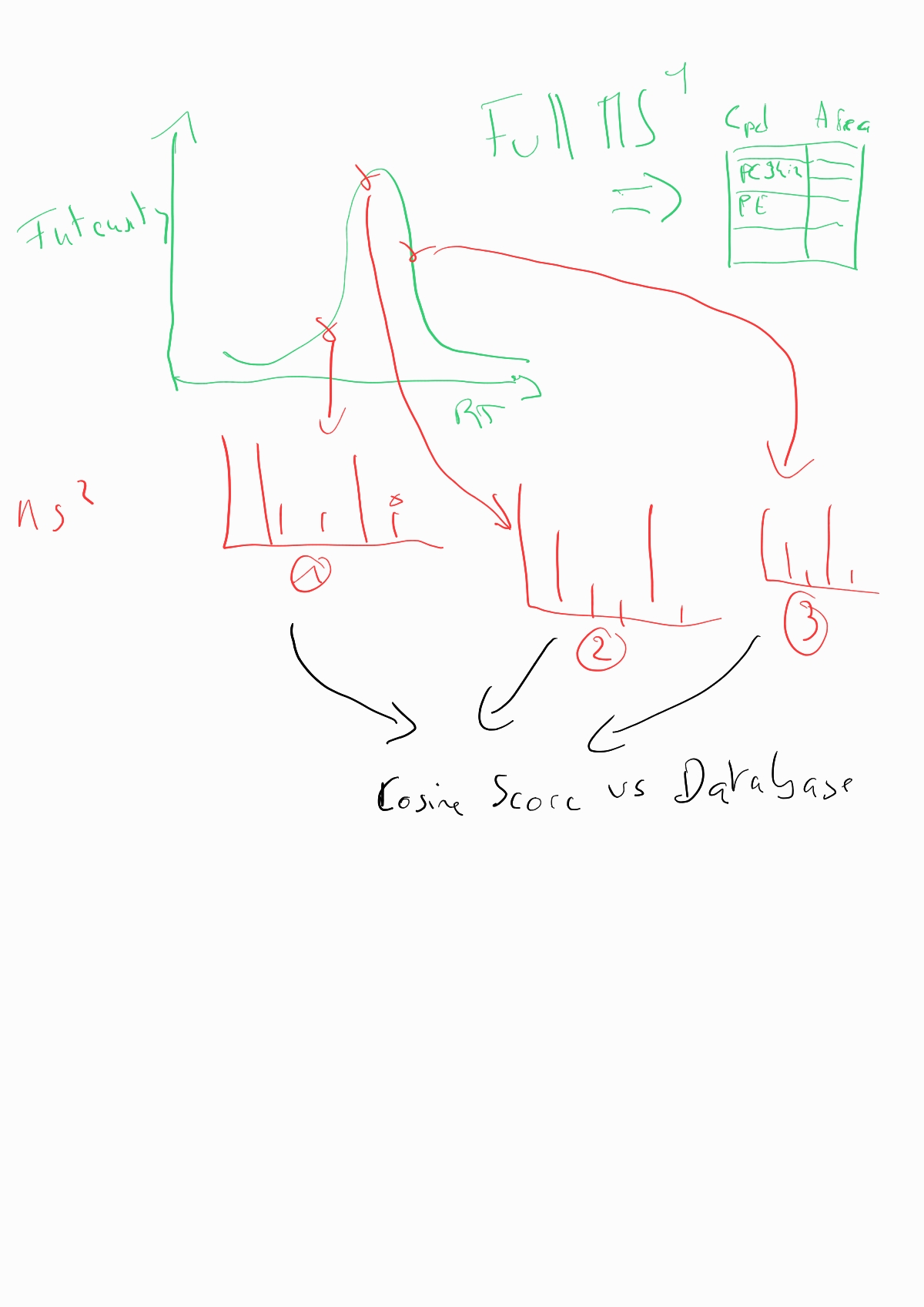
Dear Skyline support, We have a global lipid analysis method on a Thermo Exploris 240. We analyse in DDA. We have molecules with identical monoisotopic masses that we can differentiate thanks to their fragmentation. That's why we always look at our fragmentation spectra to confirm the molecules. We are currently reprocessing with Tracefinder software. On Skyline, I've tested it with ‘molecule interface’, with a list of compounds of interest. The areas are integrated in full scan, but I don't have MS/MS visualisation. I tested Lipid creator. Theoretical fragmentations are created, with a ‘Library Match’ display with the expected fragments and red and green colour codes in the list of molecules to confirm the fragments detected. But I think the integration is only done on the MS/MS triggered. And if not enough MS/MS are triggered, the peaks are not Gaussian. That's why I'd prefer integration on the FS, while viewing the MS/MS under each peak. My question is: Is it possible on Skyline to integrate the FS while displaying the MS/MS for small molecules? Thank you for your consideration. Best regards Océane DELOS |
|||||||||||||||||||||||||||
| |||||||||||||||||||||||||||
 Notes_250317_183931.jpg
Notes_250317_183931.jpg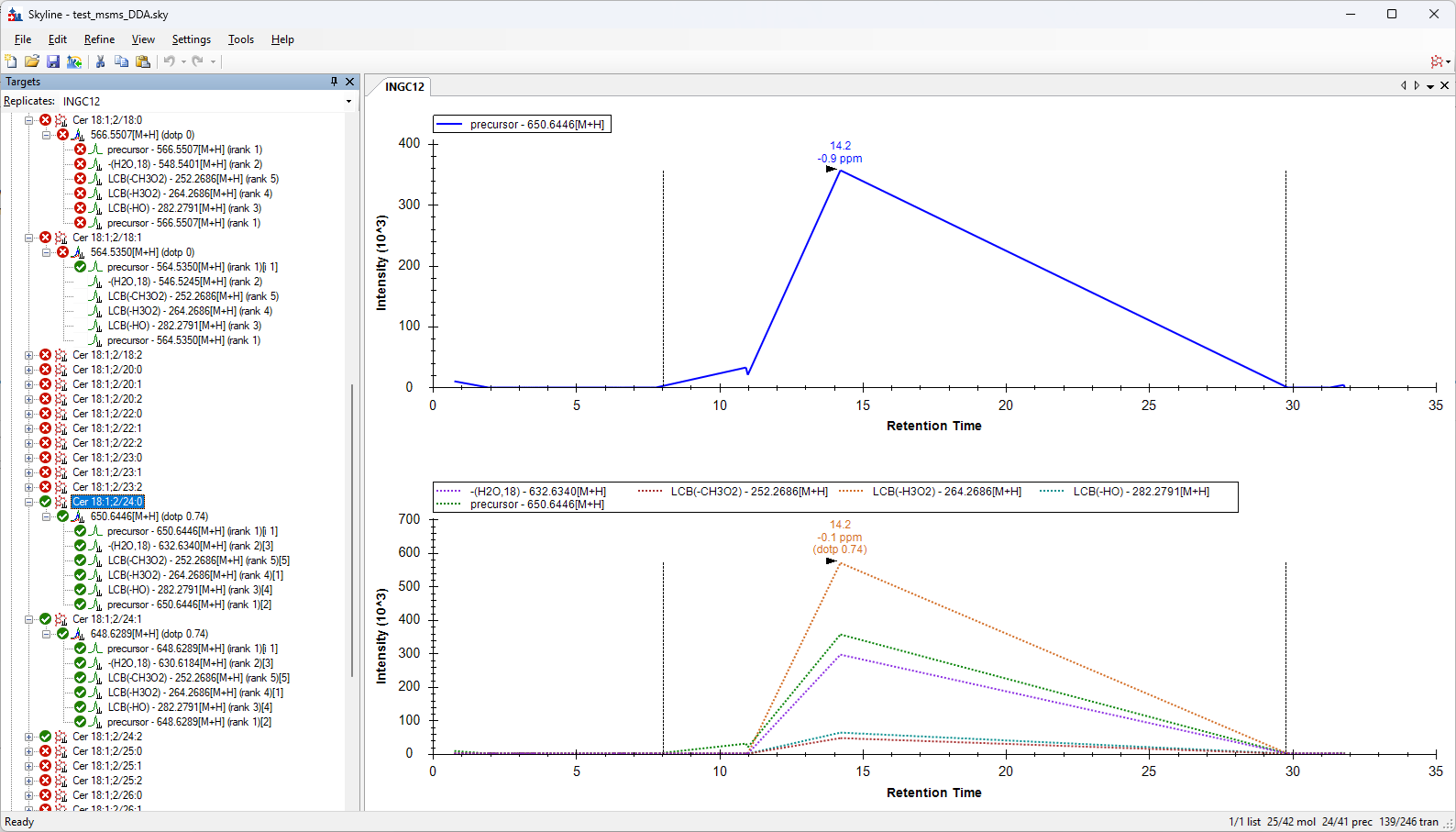 Chromatogram.png
Chromatogram.png