| |
| Brian Pratt responded: |
2025-03-04 13:25 |
It looks fine, other than I would not place the labeled precursor in a different Molecule List. Skyline needs to understand that it is related to the unlabeled version. I just copied the cells in your spreadsheet, except for the first column, and pasted that into Skyline and it looks good.
Have you had a look at https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_small_quant ?
Thanks for using the Skyline support board,
Brian Pratt
|
| |
| Nick Shulman responded: |
2025-03-04 13:30 |
Here is the transition list that I would use.
Your heavy labeled standard should have the same "Molecule List Name" and "Molecule Name" as the light molecule.
In your Excel spreadsheet, the heavy 5-Methyl THF had a different Molecule List Name, so Skyline would end up thinking they were completely unrelated molecules.
What type of mass spectrometer do you have?
If you acquired SRM chromatograms on a triple quadrupole mass spectrometer then, after you have inserted your transition list, you can tell Skyline to read the chromatograms using the menu item "File > Import > Results".
If your data was something else such as PRM then you will need to go to the "Full Scan" tab at "Settings > Transition Settings" and tell Skyline your MS/MS acquisition method.
We actually have a tutorial specifically about small molecule calibration curves:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_small_quant
If you get stuck anywhere, you should send us your Skyline document.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
Files which are less than 50MB can be attached to these support requests. You can always upload larger files here:
https://skyline.ms/files.url
-- Nick |
|
| |
| weitmanm responded: |
2025-03-04 17:38 |
Wow, that was amazing! Your support made a huge difference!
I’m working with an Orbitrap instrument, and the combination of adjusting the settings along with correcting the transition list file completely changed my results. Before this, I wasn’t able to detect the fragment ion properly.
Thank you so much for directing me to the small molecule tutorial. I’ll follow the guide and, if I have any further questions, I know I’m in great hands!
From a quick look, when I exported the peak areas to a CSV report, I noticed that each molecule has two columns. I assume one corresponds to the precursor ion and the other to the fragment ion. To be more specific, I believe I should focus on the fragment ion area since it combines both precursor and fragment information, which is crucial in a biological background, right?
Thanks again for the outstanding support!
Michal |
| |
| Nick Shulman responded: |
2025-03-04 18:45 |
|
| |
| weitmanm responded: |
2025-03-05 12:44 |
Thanks for the report tutorial!
I encountered an issue while setting up my calibration curve.
I prepared the calibration curve using a heavy-labeled compound. Since I wanted to perform the calibration in the experiment matrix—and my target compound is always present in the biological matrix across all groups—I prepared spikes at different concentrations using the heavy-labeled compound.
After defining the samples that belong to the calibration curve, Skyline does not compute the calibration curve for the heavy compound, and I receive the following error message:
"Error: All of the external standards are missing one or more peaks."
How can I resolve this issue?
Thanks! |
| |
| Nick Shulman responded: |
2025-03-05 15:41 |
One thing that could cause that "All of the external standards are missing one or more peaks" is if Skyline thinks everything is an internal standard.
That is, you should go to the "Labels" tab at "Settings > Molecule Settings" and uncheck the box next to "heavy".
The problem is that Skyline expects the internal standard to be present in all samples at the same quantity, so, if all Skyline sees for a molecule are internal standards, Skyline gives you the confusing message that peaks are missing but what it really means is that it could not figure out which transitions to use.
If that does not fix the problem then you should send us your Skyline document using the "File > Share" menu item to create a .zip file and either attaching it this support request or uploading it to https://skyline.ms/files.url
-- Nick |
| |
| weitmanm responded: |
2025-03-06 06:54 |
Wow, Nick, thank you! That worked like magic.
I have a few follow-up questions:
1. Normalization Approach
In my experiment, I:
a. Spiked a fixed concentration of the heavy-labeled compound into all biological samples with unknown concentrations.
b. Created a calibration curve using the heavy-labeled compound in the same biological matrix.
To normalize my data, I was considering the following approach:
I. Calculate the average signal of the heavy-labeled compound across all samples.
II. Compute the ratio between the measured heavy-labeled compound in each sample and this average.
III. Use this ratio as a normalization factor.
Normalize the light compound’s signal by dividing it by this factor.
IV. Use the calibration curve to determine the absolute concentration of the unknown compound in each sample.
Is this approach correct?
Are there any factors I might not have considered that could affect accuracy?
Is there a way to implement something similar within Skyline, or should I export the results to Excel and perform the calculations there? Alternatively, do you have a different approach that you would recommend?
2. Missing Parent Ion Chromatogram
One of the metabolites I’m tracking is folic acid.
In some samples he chromatogram for the parent ion appears empty.
However, I do see a chromatogram for the fragment ion.
I’ve attached a screenshot. Does this seem reasonable to you?
Thanks again for your insights—I really appreciate your help!
Michal |
|
| |
| Nick Shulman responded: |
2025-03-06 08:29 |
Can you send us your Skyline document and your raw data?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
The Share Document dialog gives you the option of including the raw data files in the .zip, or you could zip those up separately and send them to us.
Files which are less than 50MB can be attached to these support requests.
You can always upload larger files here:
https://skyline.ms/files.url
I do not understand your description of your plan for normalizing the data. Skyline knows how to do normalization by dividing the light signal by the heavy signal. You tell Skyline that you want to normalize to heavy either by choosing "Ratio to Heavy" as the Normalization Method at "Settings > Molecule Settings > Quantification" or by using the Document Grid to set the Normalization Method of the individual molecule.
Sometimes people might want to multiply by the average heavy area across all of the samples after they have divided by particular heavy area. They might do this because they want the normalization factor to be close to 1 so that the measured light area and the normalized light area would be close in magnitude to each other. But most people are perfectly happy to have the normalized area be the ratio of light to heavy which is what Skyline can easily tell you.
After I see your Skyline document and raw files I might be able to answer your question about the parent ion chromatogram.
-- Nick |
| |
| weitmanm responded: |
2025-03-06 08:55 |
Thank you, Nick!
I have uploaded the files, and I would appreciate it if you could check that I did it correctly.
I will review what you wrote regarding normalization.
Thanks a lot for now! |
| |
| Nick Shulman responded: |
2025-03-06 13:39 |
Thank you for uploading that Skyline document.
Can you send me the file "20250226_QEF1_C18_MC38OVA_folates_5me-THFlabelled_KH31_TIF.raw"?
In general, if you want to know why a particular point along a chromatogram has the intensity that it does, you can click on that point in the chromatogram graph in Skyline.
Skyline will try to find that .raw file and will show you the spectrum that contributed to that point in the extracted ion chromatogram.
Skyline will also highlight on the spectrum the m/z range around the transition m/z that Skyline summed across to extract the intensity.
If you see that there is some signal just outside of the window that Skyline was summing across, you might be able to fix things by lowering the resolution at "Settings > Transition Settings > Full Scan".
After you make a change like that you will need to tell Skyline to extract chromatograms again by using the "Reimport" button at "Edit > Manage Results".
By the way, for Thermo instruments we usually recommend that you choose "Centroided" as the mass analyzer on the "Full Scan" tab at "Settings > Transition Settings".
The reason for this is that Thermo's centroiding algorithm does a better job of separating signal from molecules with similar m/z so you can get a cleaner signal than what you get if Skyline does the work of summing the signal from a profile spectrum.
-- Nick |
| |
| weitmanm responded: |
2025-03-10 13:37 |
Hi Nick,
Thank you so much!
I’ll start with the last point—
Would you recommend setting "centroid" in the Full Scan settings instead of "Orbitrap"? Or, since I’ve already defined the instrument as an Orbitrap, is there an additional place where I should adjust this setting?
Secondly, the file you requested—I’m attaching it here, along with a file containing information about the folic acid standard (see folateMix file). In that file, you can clearly see the chromatogram for the precursor ion.
However, in the experimental files, I am unable to see the precursor ion…
I am indeed verifying that my chromatogram contains the correct peak and that the isotopic pattern in MS1 matches expectations, that's why I am bit confused with the folic acid results...
Looking forward to your insights! |
| |
| weitmanm responded: |
2025-03-10 13:38 |
Hi Nick,
Thank you so much!
I’ll start with the last point—
Would you recommend setting "centroid" in the Full Scan settings instead of "Orbitrap"? Or, since I’ve already defined the instrument as an Orbitrap, is there an additional place where I should adjust this setting?
Secondly, the file you requested—I’m attaching it here, along with a file containing information about the folic acid standard (see folateMix file). In that file, you can clearly see the chromatogram for the precursor ion.
However, in the experimental files, I am unable to see the precursor ion…
I am indeed verifying that my chromatogram contains the correct peak and that the isotopic pattern in MS1 matches expectations, that's why I am bit confused with the folic acid results...
Looking forward to your insights! |
| |
| Nick Shulman responded: |
2025-03-10 14:11 |
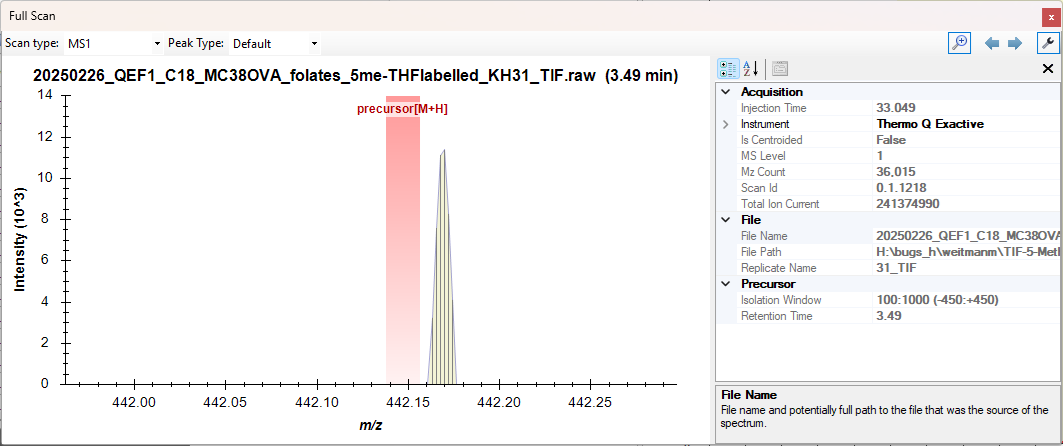
Thank you for uploading that .raw file.
When I clicked on a point along that flat precursor chromatogram, I saw the attached image.
Skyline shows a highlighted region around the precursor m/z which is the channel that Skyline summed across when extracting that chromatogram point.
You can see that there is some signal just outside of that signal so if you were to choose a lower resolution value at "Settings > Transition Settings > Full Scan" and then reimported results, you would see a non-zero intensity on the chromatogram graph.
If you changed the mass analyzer to "Centroided" and chose a mass accuracy of "60" and then did a reimport, you would see a non-zero intensity for the precursor there.
It's possible that the signal in that screenshot is not coming from your molecule of interest because having to use "60" as the mass accuracy might be unreasonable depending on how well you believe your instrument was calibrated.
Alternatively, you could leave the mass analyzer as "Orbitrap" and change the resolution value to something like "15,000".
The reason that Skyline asks you what type of instrument it is on the Full Scan tab is to figure out what to do with the resolution number so that Skyline knows how wide of a m/z channel to sum across when extracting chromatograms.
The QIT resolution can be represented by a single delta m/z value. Also, the QIT is considered low-resolution so Skyline will not extract separate MS1 chromatograms for each isotope mass.
The resolution of a TOF is proportional to the m/z so you only have to provide a single number which is the reciprocal of the parts per million.
The Orbitrap and FT-ICR have resolutions which are based on two different complicated formulas so you have to tell Skyline a resolution value and the m/z value at which that resolution applies and Skyline does the math to figure out what the resolution is at different values.
-- Nick |
|
| |
| weitmanm responded: |
2025-03-10 17:28 |
Got it!!
Based on all this, it seems that these samples probably don’t contain folic acid, since there’s no reason for the exact mass to drop so dramatically—especially when I know that other compounds at the same mass range were measured well. Plus, the folate mix contains folic acid, and it appears clearly there.
Thanks so much for the detailed explanation! |
| |
|
|
 Spectrum.png
Spectrum.png