Peptide search results should not be used to infer the absence of a peptide.
That is, you might say "Proteome Discoverer did not find the peptide in this sample" but that is not the same as "Proteome Discoverer said that this peptide is not present in this sample".
It is likely that Proteome Discoverer found rather good evidence of the peptide in that sample, but the evidence was not strong enough to be reported with a 1% false discovery rate cutoff.
Once a peptide has been detected in one of your samples, you can assume that the peptide is present in all samples, although it might be present at an amount that is below the limit of detection.
If you integrate the chromatogram peak area at the place where you expect the peptide to be found, you can get an upper bound on the quantity of peptide that might be present.
If you are interested in comparing quantities between groups of samples, and determining which peptides are present in different amounts in the two groups, we recommend the Group Comparison tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_grouped
-- Nick
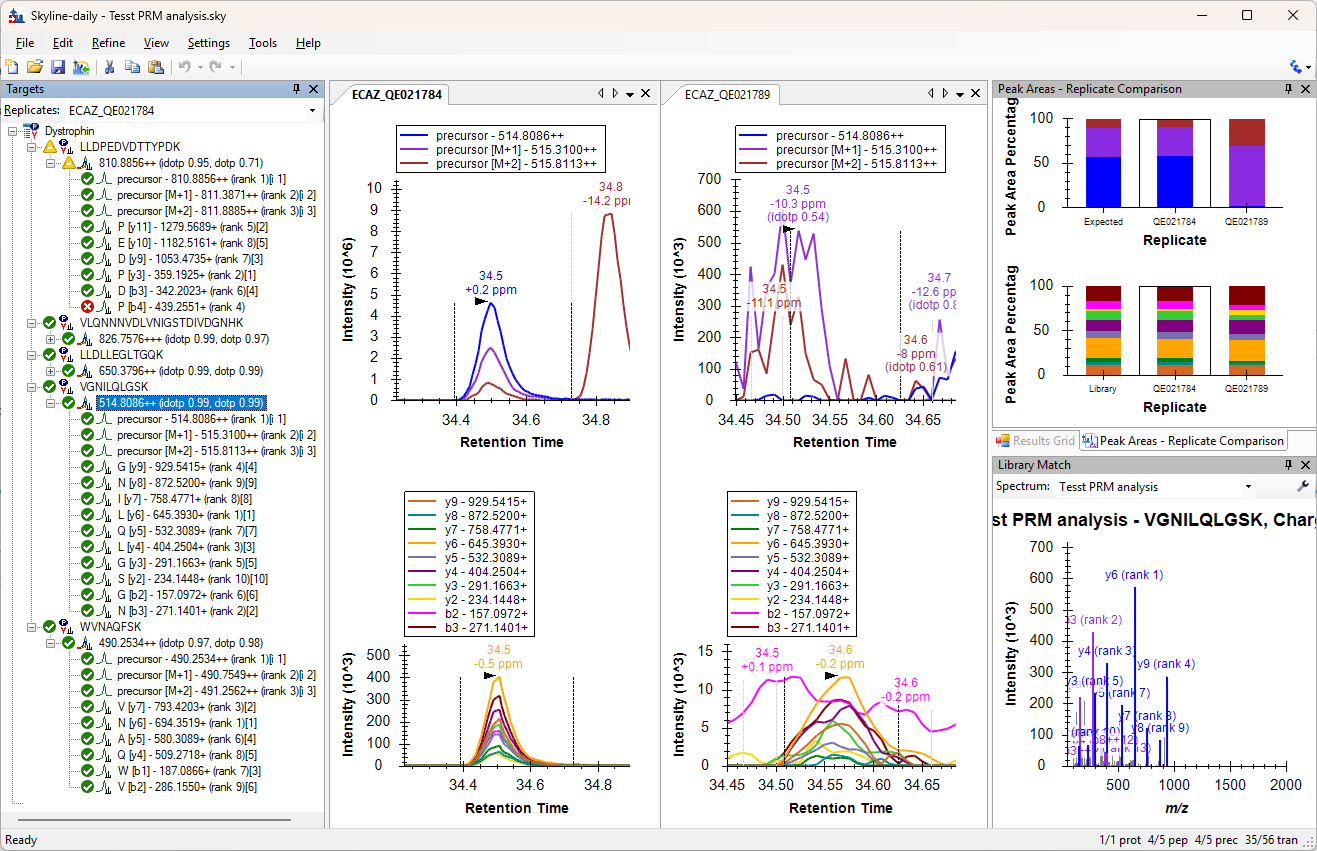 SkylineScreenshot.png
SkylineScreenshot.png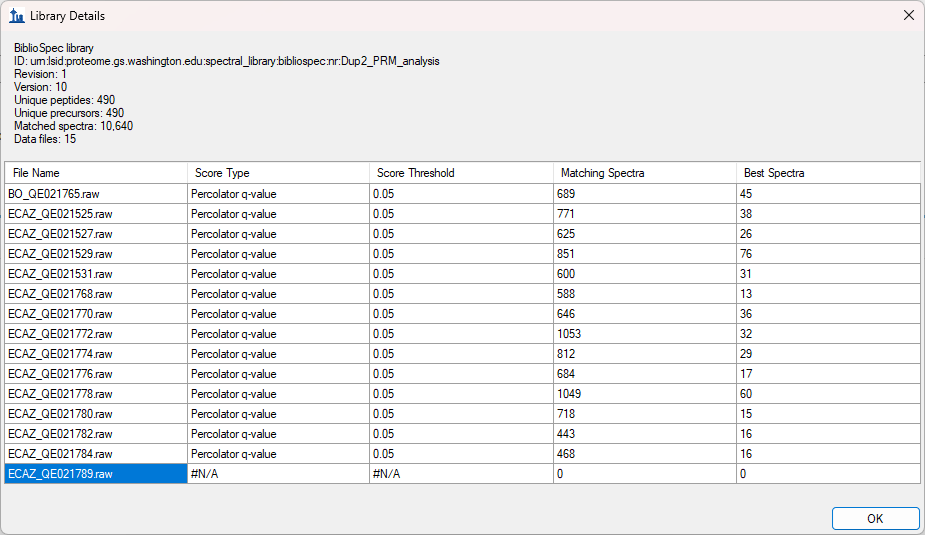 LibraryDetails.png
LibraryDetails.png