| Nick Shulman responded: |
2024-11-19 22:05 |
I do not know the answer to your question but if you send us your raw files and your FASTA file we could take a look.
You can upload all those files here:
https://skyline.ms/files.url
-- Nick |
| |
| vanyabangera responded: |
2024-11-20 02:02 |
Hello Nick,
Thank you for your response.
I have uploaded a file named "GPF_runs_no_matches_passed_score_filter_in". IKindly look into this and let me know.
Thank you
Vanya |
| |
| Nick Shulman responded: |
2024-11-20 08:25 |
Thank you for uploading those .raw file and that FASTA file.
If you changed the "Max q-value" setting to a higher number then I believe you would get some matched peptides and you would be able to successfully complete the Run Peptide Search wizard.
The "Max q-value" setting is on the "Search Settings" page of the wizard and by default its value is 0.01 but you could change that to 0.1.
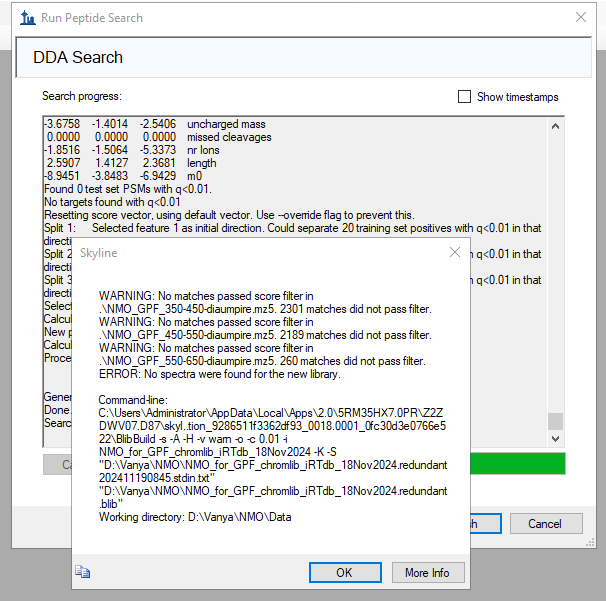
When I do that, Skyline does manage to find a few peptides about 200 peptides, which is probably much less than would be expected for a sample like this.
I think the next step would be to choose a peptide which you know is easy to detect in the sample, go find the spectra that it would be expected to be in and see what those spectra look like.
Did you samples contain any standard peptides that were spiked in and would be easy to find?
I would recommend using the "Edit > Insert > Peptides" menu item to add these peptides to the Skyline document and then use the menu item "File > Import > Results" to tell Skyline to extract chromatograms.
Then, click on points along the MS2 chromatogram where you think the peptide is and see whether the spectra contain the transitions you would expect.
A common reason that no peptides might be found could be that the instrument was not calibrated correctly so the transition m/z's are too far from their correct value for the peptide search engine to consider it a match. This sort of problem can be corrected by choosing a higher mass accuracy value on the Search Settings page of the Run Peptide Search wizard.
-- Nick |
|
| |
| vanyabangera responded: |
2024-11-20 20:10 |
Hello Nick,
Thank you so much for your response.
To answer your question on standard peptides, I have spiked PierceTM Peptide Retention Time Calibration Mixture at a concentration of 200fmol.
Thank you
Vanya |
| |
| Nick Shulman responded: |
2024-11-20 22:15 |
Those heavy PRTC peptides are very easy to find and their chromatograms look great, so I don't think there's anything wrong with the calibration of your instrument.
I do not know very much about how to actually use a mass spectrometer, so I do not know what would be a good idea to check next.
One thing I did notice is that MS2 spectra in the file "NMO_GPF_450-550.raw" that you uploaded have precursors in the range from 350-450 which does not seem right.
-- Nick |
|
| |
| vanyabangera responded: |
2024-11-20 22:56 |
Hello Nick,
Thank you for being so supportive. I might have gone wrong and I will look into this.
Thank you
Vanya |
| |
| vanyabangera responded: |
2024-11-20 23:33 |
Hello Nick,
Upon reviewing the raw files based on your feedback, I noticed a discrepancy in the isolation window for GPF-450-550. To address this, I will proceed with reacquiring the samples. Thank you for your valuable suggestions and guidance.
Vanya |
| |
 No_matches.PNG
No_matches.PNG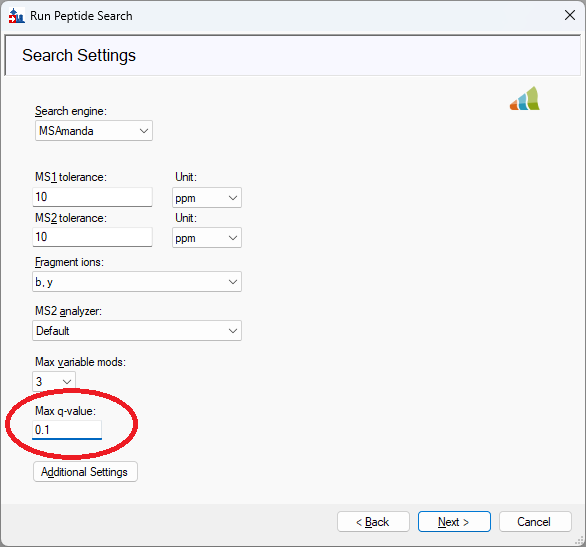 SearchSettings.png
SearchSettings.png