| Nick Shulman responded: |
2024-10-14 06:44 |
Are you seeing some sort of error message?
If you send us your files we can try to figure out what is going wrong.
I am not sure what files we would need but maybe you could package everything into a .zip file and upload it here:
https://skyline.ms/files.url
--Nick |
| |
| JeromeVia responded: |
2024-10-17 04:01 |
I upload just now
file name
Hela_DiagonalPASEF
I also add the reference skyline wirh DiaPASEF method
Hela_diaPASEF |
| |
| Nick Shulman responded: |
2024-11-05 16:38 |
Thank you for uploading those files.
I am not sure what I am supposed to be looking at.
Can you post a screenshot of what you are seeing and describe what you were hoping it would look like?
-- Nick |
| |
| JeromeVia responded: |
2024-11-08 02:10 |
Hi
I uploaded bruker data with DiagonalPASEF acquistion method. You could play with it and see what's wrong
File name: JeromeVia_241108.rar
Jerome |
| |
| Nick Shulman responded: |
2024-11-08 10:36 |
Thank you for uploading those Bruker .d data folders.
Those are slightly different runs than were in your original Skyline document "Hela_DiagonalPASEF.zip" but I think they are showing the same characteristic jagged chromatograms like in the attached screenshot "JaggedChromatogram.png".
If you click on a point along the chromatogram Skyline will show you the spectrum which contributed to that extracted chromatogram point. You can click the wrench button to show the properties.
There are many points on the chromatogram which have exactly the same retention time but slightly different intensities. These points come from spectra that have slightly different ion mobility values and also slightly different isolated precursor m/z's.
Usually when extracting chromatograms from ion mobility data, spectra with identical retention times get combined into a single spectrum. This combine spectrum has an array of ion mobility values along with the usual m/z and intensity arrays. In this way, Skyline is able to perform ion mobility filtering on this single spectrum.
However, in your data, these spectra with identical retention times are not being combined.
I am not the expert on ion mobility chromatogram extraction in Skyline, but I imagine that the reason that these spectra are remaining separate is that they have slightly different isolated precursor m/z's.
I will have to ask my coworkers what is supposed to be done with this sort of data. It is possible that this "diagonal" sort of acquisition method is not something that is supported in Skyline yet.
-- Nick |
|
| |
| Brendan MacLean responded: |
2024-11-08 11:31 |
It is not yet supported. We were unable to continue development for Bruker instruments at our normal pace this year. We are hoping to resume the effort in 2025. Let your Bruker rep know you want to see Skyline keeping up with advances in Bruker instruments and acquisition methods.
Thanks for posting this request.
—Brendan |
| |
| JeromeVia responded: |
2024-11-12 01:17 |
Thank you
I will wait 2025 :-)
Jerome |
| |
 JaggedChromatogram.png
JaggedChromatogram.png Spectrum1.png
Spectrum1.png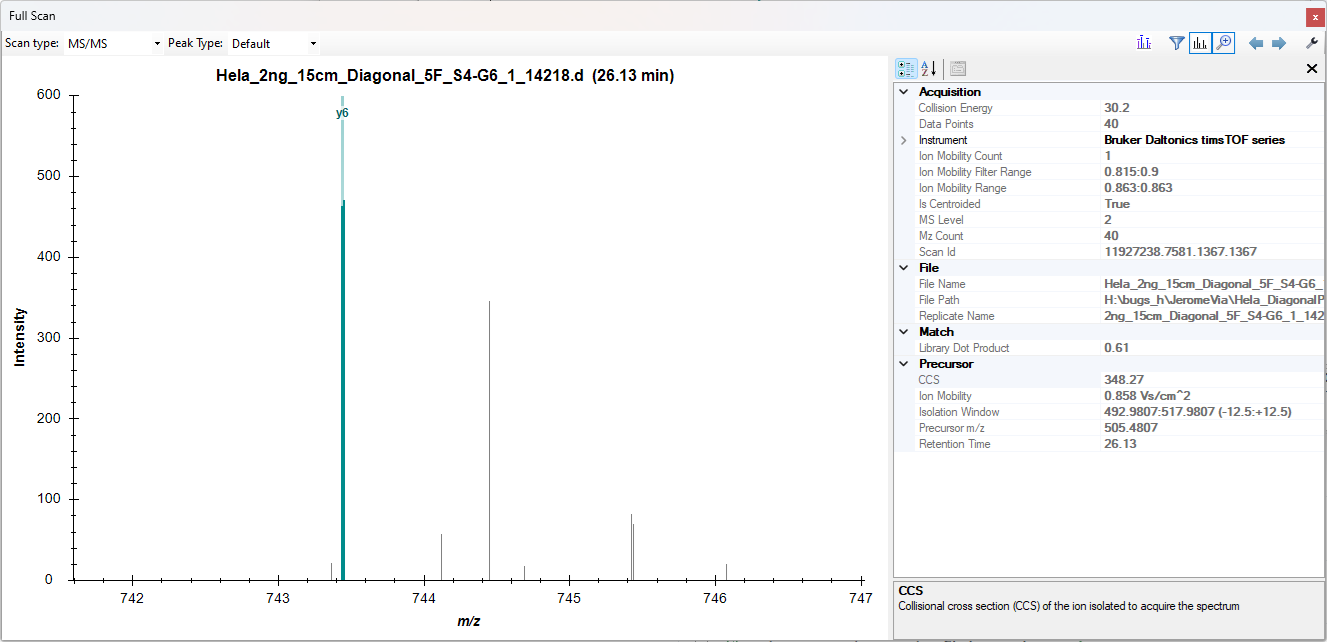 Spectrum2.png
Spectrum2.png