| Nick Shulman responded: |
2024-10-01 12:52 |
When you use the "File > Import > Peptide Search" menu item in Skyline, Skyline uses a program called "BiblioSpec" to convert the peptide search results into a spectral library.
This is the page which lists all of the peptide search result formats that BiblioSpec understands:
https://skyline.ms/wiki/home/software/BiblioSpec/page.view?name=BlibBuild
The page says that for MSFragger you would need .pepXML or .pep.xml files as well as the mass spec data files (.raw, .mzML etc).
-- Nick |
| |
| afshari1 responded: |
2024-10-06 17:35 |
Thanks for your response.
I created the spectral library through this way: Peptide Settings >> Library. I only imported mod.pep.xml files, and Skyline did not ask me for mass spectrometry data files (.raw, .mzML, etc.). I am wondering if I have created the spectral library correctly. |
| |
| Nick Shulman responded: |
2024-10-06 18:02 |
After you have created your spectral library, you can use the menu item:
View > Spectral Libraries
to see what is in the library.
It sounds like you did create your .blib file correctly because Skyline did not tell you anything went wrong.
When you build a spectral library from the Peptide Settings, you do not get asked where to find the spectrum files. BiblioSpec is able to find those files if they are in the same folder as the pep.xml files, or in a couple of other possible related folders such as the parent folders.
If BiblioSpec is not able to find the spectrum files then you will get an error.
Now that you have a spectral library, you probably want to add some peptides to your Skyline document and then tell Skyline to extract some chromatograms.
There are many different ways to add peptides to your Skyline document.
One way is to go to "View > Spectral Libraries" and push the "Add" or "Add All" buttons.
A different way is to use the "File > Import > FASTA" menu item.
After you have added peptides you can tell Skyline to extract chromatograms with the "File > Import > Results" menu item.
The usual way to build a library, add peptides, and extract chromatograms is with the "File > Import > Peptide Search" menu item.
But it is also valid to do these steps separately.
-- Nick |
| |
| afshari1 responded: |
2024-10-06 18:29 |
Thanks.
Actually, to build an unscheduled PRM method, I followed these steps:
I inserted the sequences of peptides of interest (100 targets) through the edit menu.
Then, I exported the report file to set up the method on the instrument.
Now, I have created the spectral library and imported my PRM results (10 runs) into Skyline to perform data analysis.
I hope I have done the steps correctly so far. However, when I select some of the peptides in my target list, on the chromatograms, I get two messages:
Sometimes it says "Select a peptide or precursor to view its chromatogram" even though I have already selected the peptide.
Other times it says "Chromatogram information is unavailable."
I am not sure what these messages mean. Please see the attached files. |
|
| |
| Nick Shulman responded: |
2024-10-06 18:50 |
The "Chromatogram Information Unavailable" message usually means that no chromatograms were extracted for the currently selected peptide.
For a PRM experiment like this, it basically means that there were no MS2 spectra whose isolation window matched the m/z of the precursor in your Skyline document.
If you send us your Skyline document and your .raw files we can tell you why you are not getting chromatograms.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including spectral libraries and extracted chromatograms.
The Share Document dialog gives you the option to include the .raw files, or you could send them to us separately.
Files which are less than 50MB can be attached to these support requests.
You can always upload larger files here:
https://skyline.ms/files.url
I see that some of your peptides have no Precursor under them. I wonder if that might have something to do with the Peptide having different modifications on it from the thing that was in the Library.
You can add precursors to your peptides by right-clicking on them in the Targets tree and choosing "Pick Children".
If you would like to learn more about modifications in Skyline, the following Webinar is helpful:
https://skyline.ms/webinar10.url
-- Nick |
| |
| afshari1 responded: |
2024-10-06 19:49 |
I just uploaded my Skyline file and raw files to the file sharing platform.
The names of the files are: raw files and unscheduled_PRM.sky.zip
Yes, these are phosphopeptides, and some of them have CAM modifications as well. Before creating the spectral library, all peptides have precursors, but when I create the spectral library, some of the precursors disappear. |
| |
| Mike MacCoss responded: |
2024-10-07 13:02 |
If you are interested in using FragPipe as input to Skyline, you can get FragPipe to generate the Skyline document for you. See attached for FragPipe screenshot. The FragPipe team has done a great job in adding features to make it easier to view quantitative data from FragPipe in Skyline.
Also did you know that you can search with MSFragger directly within Skyline? File > Search > Run Peptide Search… |
|
| |
| Nick Shulman responded: |
2024-10-07 15:00 |
afshari1,
Thank you for uploading your Skyline document and .raw files.
The reason that you see "Chromatogram Information Unavailable" for some replicates for some of your peptides is that there really are no MS2 spectra where the isolated precursor matches the m/z of the precursor in your Skyline document.
If this was a multi-injection experiment where the mass spectrometer created multiple .raw files to span the complete set of precursors, then you should tell Skyline to put multiple .raw files into a single Replicate.
When you do "File > Import > Results", Skyline asks you about whether this is a multi-injection dataset and, if so, you should choose one of the other options.
I also see that for many of your modified peptides, Skyline has given you no precursors.
One thing that you could do is right-click on one of those peptides and choose "Pick Children" and then click on the filter button to turn off the filter and then check the checkboxes next to the charge states that you want to add.
Alternatively, you could go to:
Settings > Peptide Settings > Library
and change "Pick peptides matching" to "Library or Filter".
Currently, that is set to "Library" which causes Skyline to not give you any precursors for the peptides whose modified peptide sequence is not in your library.
Hope this helps,
-- Nick |
| |
| afshari1 responded: |
2024-10-11 11:26 |
Thank you for your response.
As I am analyzing phosphopeptides, I am wondering how I can incorporate neutral loss spectra into the transition list. |
| |
| lxiiaanog responded: |
2024-12-01 23:21 |
Hi,
Following the same step, I encountered a different issue.
Skyline shows that it requires an uncalibrated.mgf file to proceed to the next step (skyline_issue.png). However FragPipe only generates an uncalibrated.mzmL file in the same directory as the raw file (filelist.png).
Is there any configuration that I wrongly set, could you help me out? Thanks!
Li |
|
| |
 chromatogram unavailable.png
chromatogram unavailable.png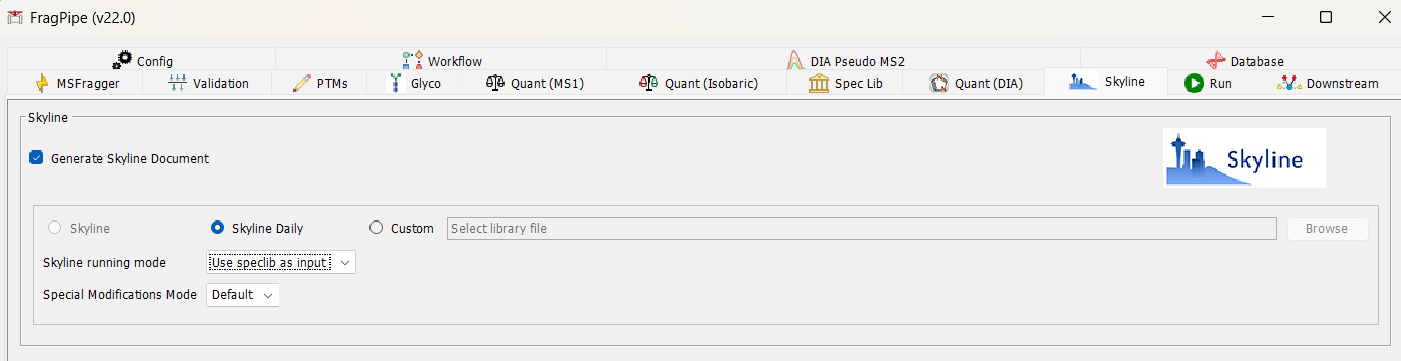 FragPipe-Skyline.png
FragPipe-Skyline.png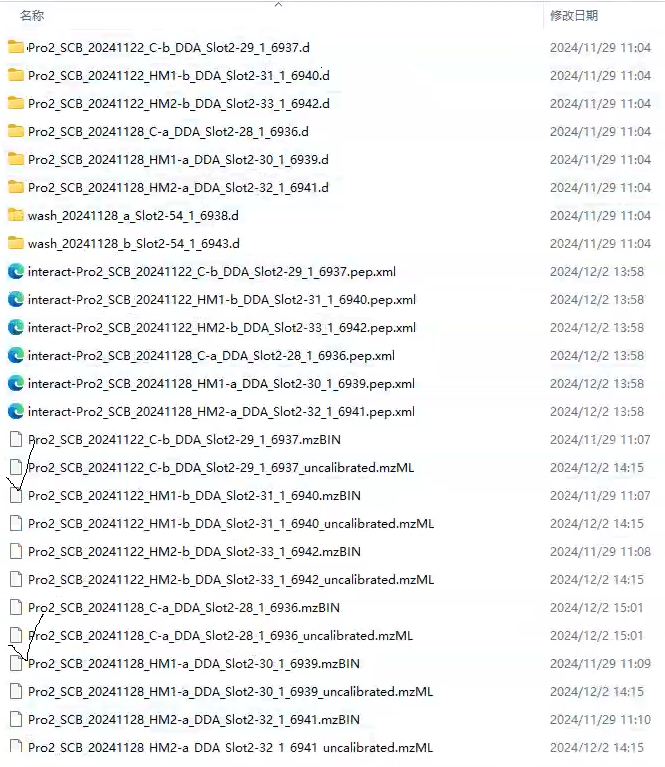 filelist.png
filelist.png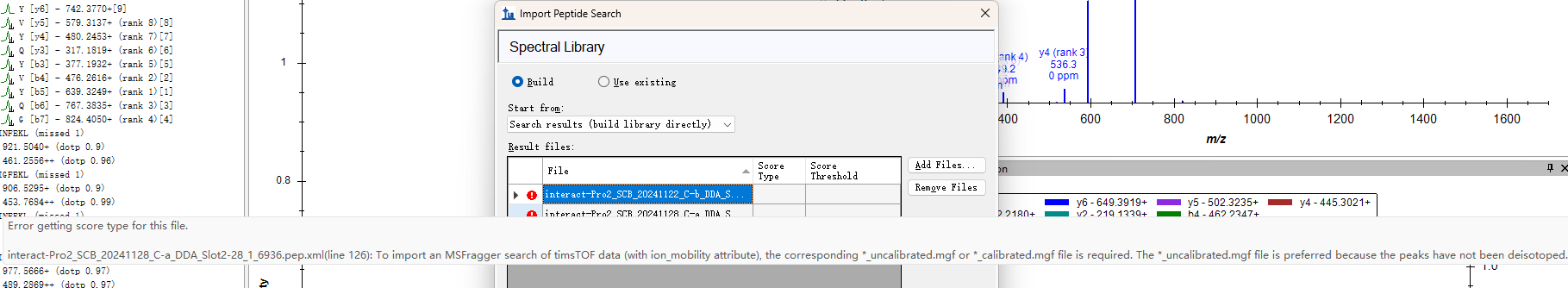 skyline_issue.png
skyline_issue.png