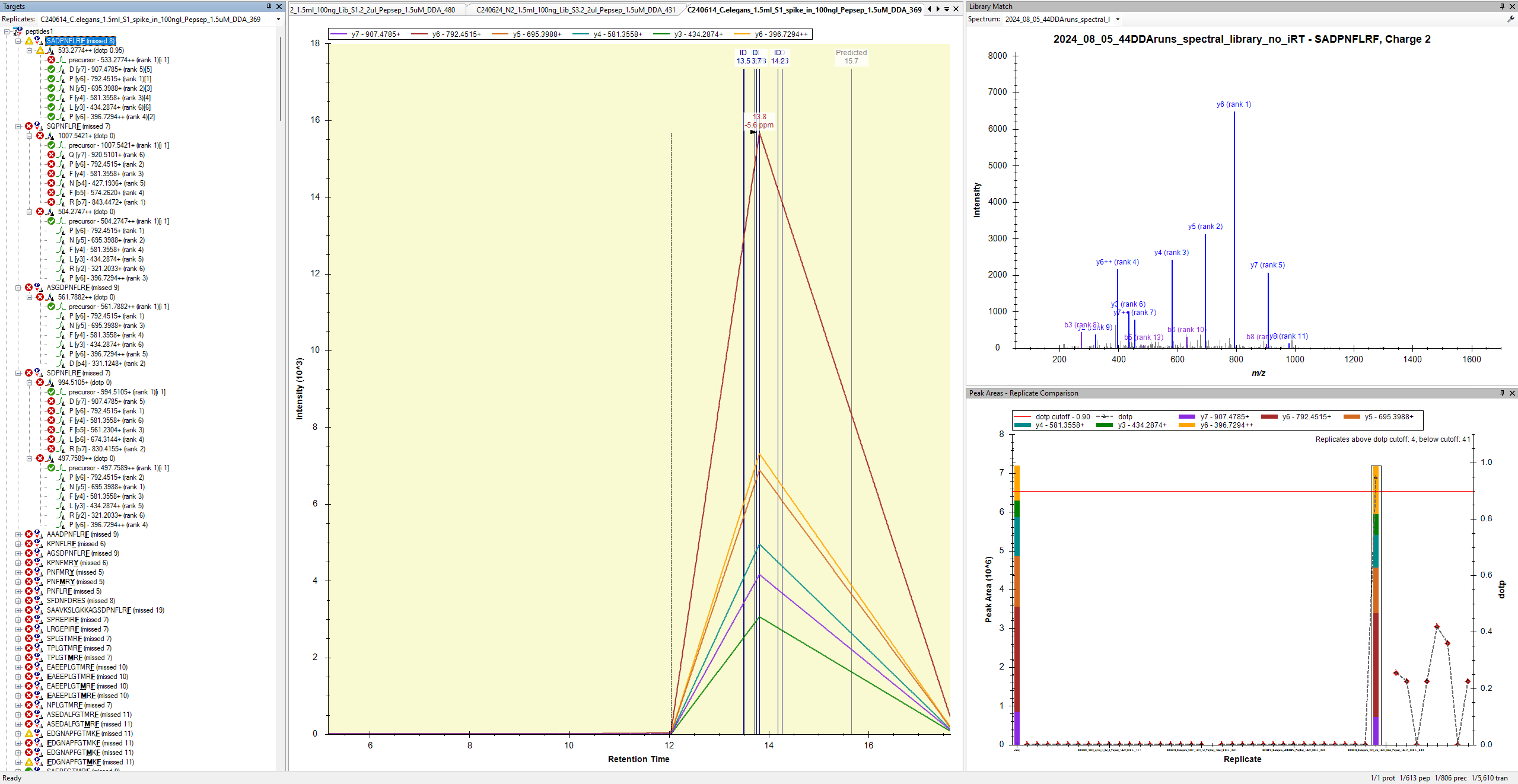
When Skyline extracts MS2 chromatograms from DDA data, the chromatograms usually look like that with long straight lines connecting the points where a matching precursor was selected for fragmentation by the mass spectrometer.
Skyline is not a good tool for looking at DDA MS2 data for this reason, because Skyline is very oriented around chromatograms.
Despite this, I am still a little confused about your screenshot because there are several ID lines on the chromatogram in a region where the chromatogram has only one extracted point. In theory, each of those ID lines represents a MS2 spectrum that your peptide search engine thought matched your peptide, so it is surprising that Skyline did not believe that there was a MS2 spectrum there from which a chromatogram point could be extracted.
It is certainly possible for that to happen, since Skyline and your peptide search engine use different criteria to decide whether a MS2 spectrum matches a precursor.
If you want to use Skyline with DDA data we recommend only using Skyline to extract chromatograms from MS1 spectra.
Skyline does allow you to extract chromatograms from DDA MS2 spectra, but, as you can see, they are not very useful.
If you are going to extract chromatograms from DDA MS2 data, be sure to choose "DDA" as the "MS/MS filtering Acquisition method" at "Settings > Transition Settings > Full Scan". If you choose either "PRM" or "DIA" then Skyline will truncated your MS1 chromatograms so that its time range matches the time range over which there were matching MS2 spectra which is not what you want. Skyline does this because it is the correct thing to do for scheduled PRM methods where the mass spectrometer has been told which precursors to monitor over which time ranges.
There are a couple of great tutorials for using Skyline with DDA data:
If you have peptide search results from another peptide search engine I recommend the "MS1 full scan filtering" tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_ms1_filtering
If you want to use one of Skyline's built-in peptide search engines, then I recommend the "DDA Search for MS1 filtering" tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_dda_search
-- Nick
 SADPNFLRF_MS2.png
SADPNFLRF_MS2.png