| Protein abundance | vmohanty | 2024-07-12 08:26 | |||||||||||||||||||||||||||||||||||
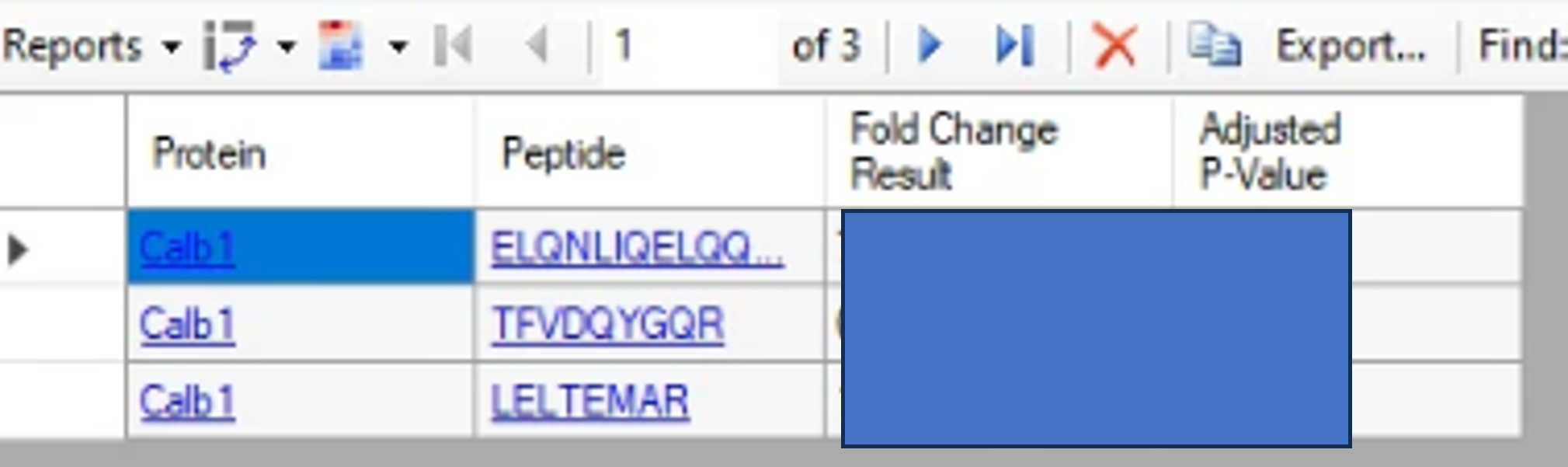
Hi All, I have acquired my PRM data and using Skyline for computing my Fold change using MS stats. I have 2 peptides corresponding to one protein and would like to know the Fold change at the protein level. I followed the Tutorial 20_1, however, my data is displayed in Fold change of each peptide with Adjusted P-Values. I am ready to share my Skyline document too. Could you please provide me with some ideas how to do so. Than would be very helpful Thank you. |
|||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
 Picture1.png
Picture1.png Prot_abundance.png
Prot_abundance.png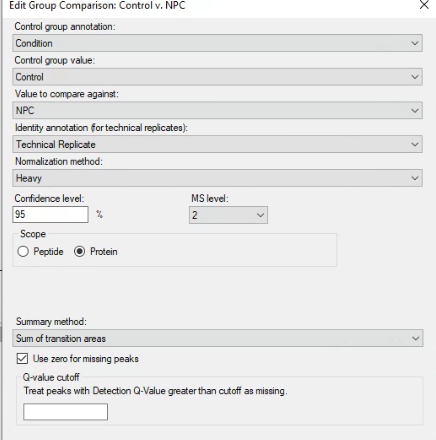 Settings.png
Settings.png