Hi,
I have a question about how Skyline is pulling transition data from Thermo .raw files and I'm going to try and attempt what I mean but I can provide a skyline file to show what I mean. Some background I'm putting in Full scan ddMS2 data from an Exploris 120.
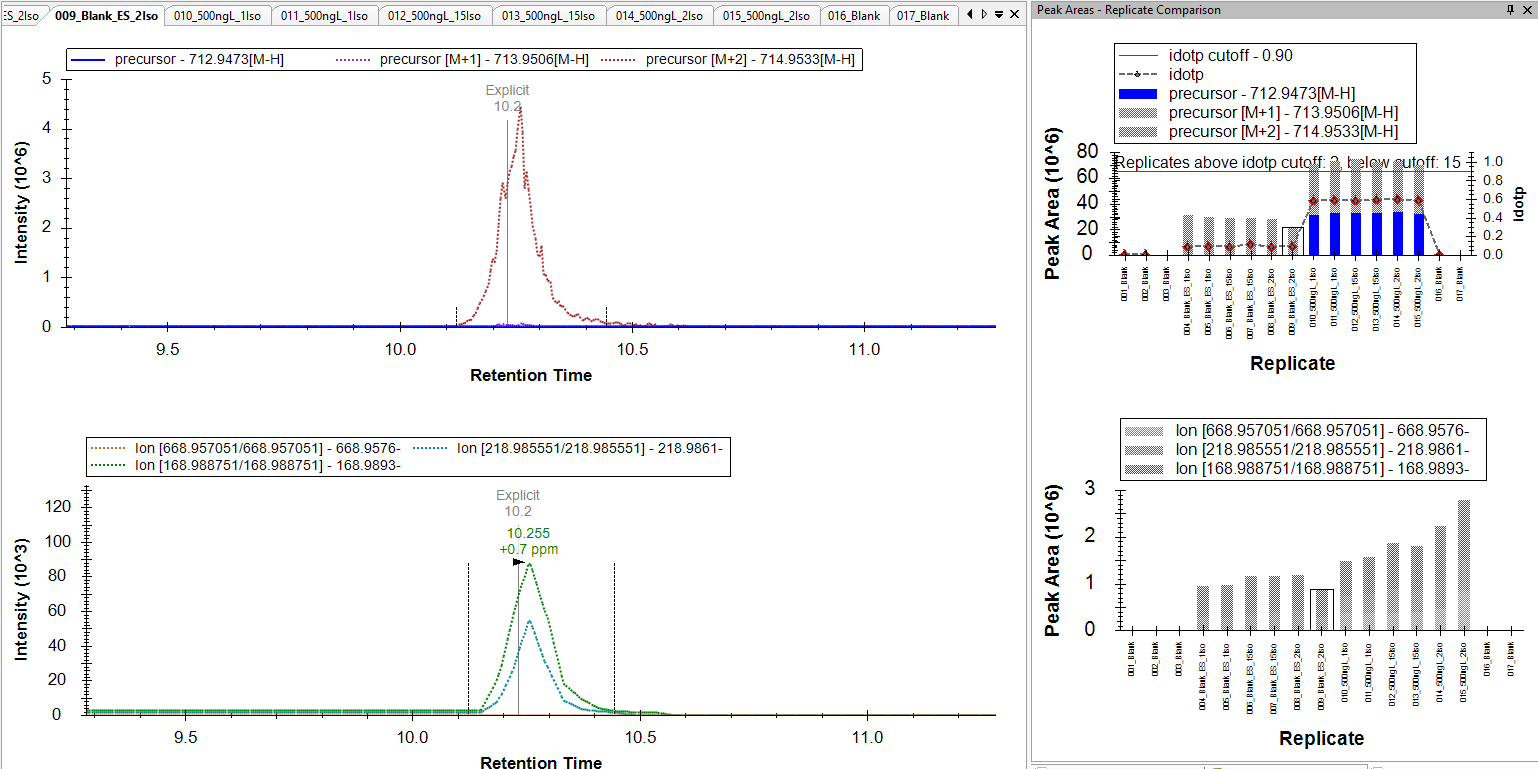
The issue we are seeing is for compounds that we have only a 2 m/z difference between the native and IS. For example, Native mass is 712.9437 and the IS is 714.9540. I've attached a picture for now to show what I mean. We have an injection for a blank and we see now peak for the native mass, but we see fragments appearing. We know the native and IS can produce similar fragments because of the low mass labeling.
So the question I have is Skyline just pulling an ion channel for transition and that means if anything produces that fragment mass at that time then it would be captured and shown? This would explain why we see fragments from the IS when there isn't any of the native present in the blank to produce those ions.
I've checked the same data in Thermo TraceFinder. When we look at the blanks there is no peak for the native and there is not fragments showing up.
 Skyline_TransitionImage.PNG
Skyline_TransitionImage.PNG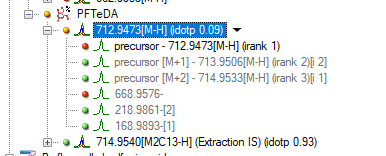 Skyline_TransitionMasses.PNG
Skyline_TransitionMasses.PNG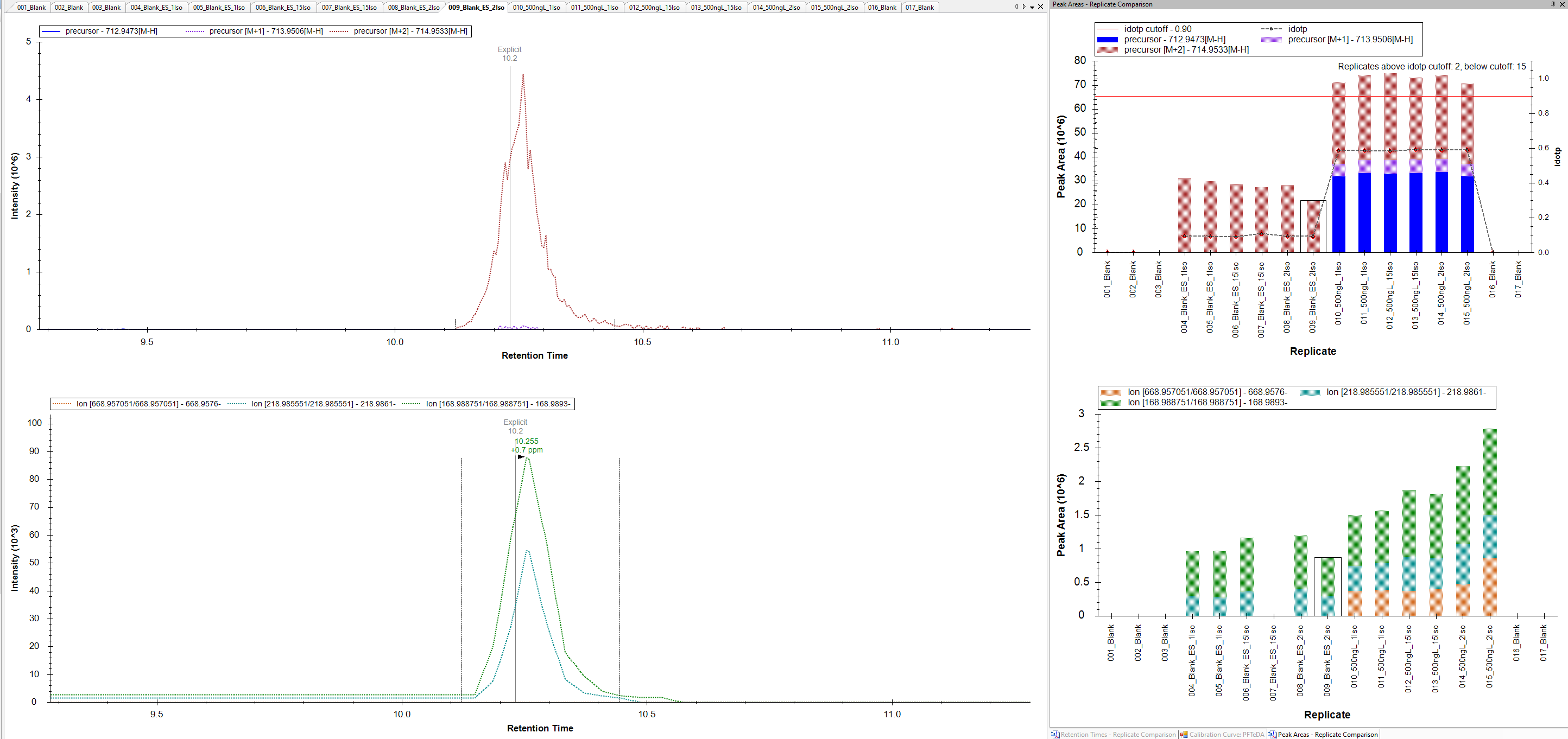 Skyline_DDA.PNG
Skyline_DDA.PNG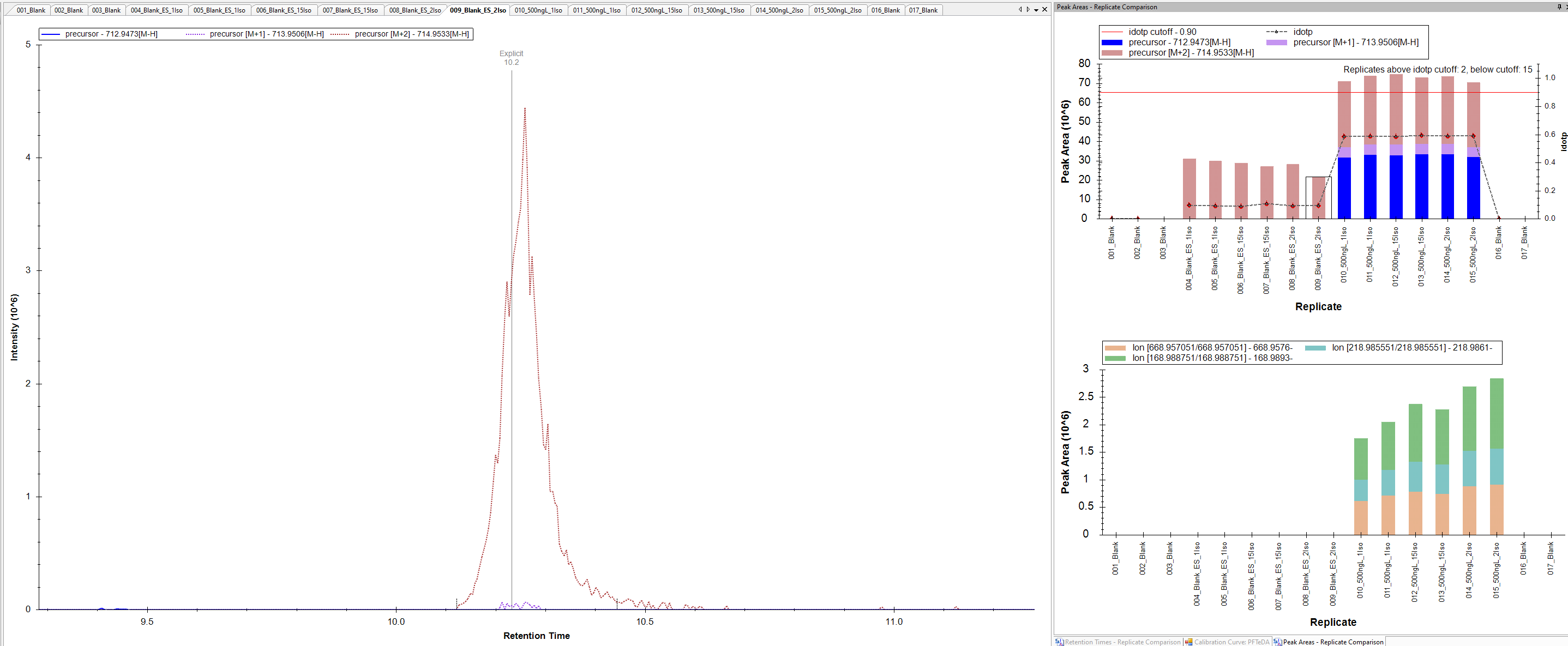 Skyline_PRM.PNG
Skyline_PRM.PNG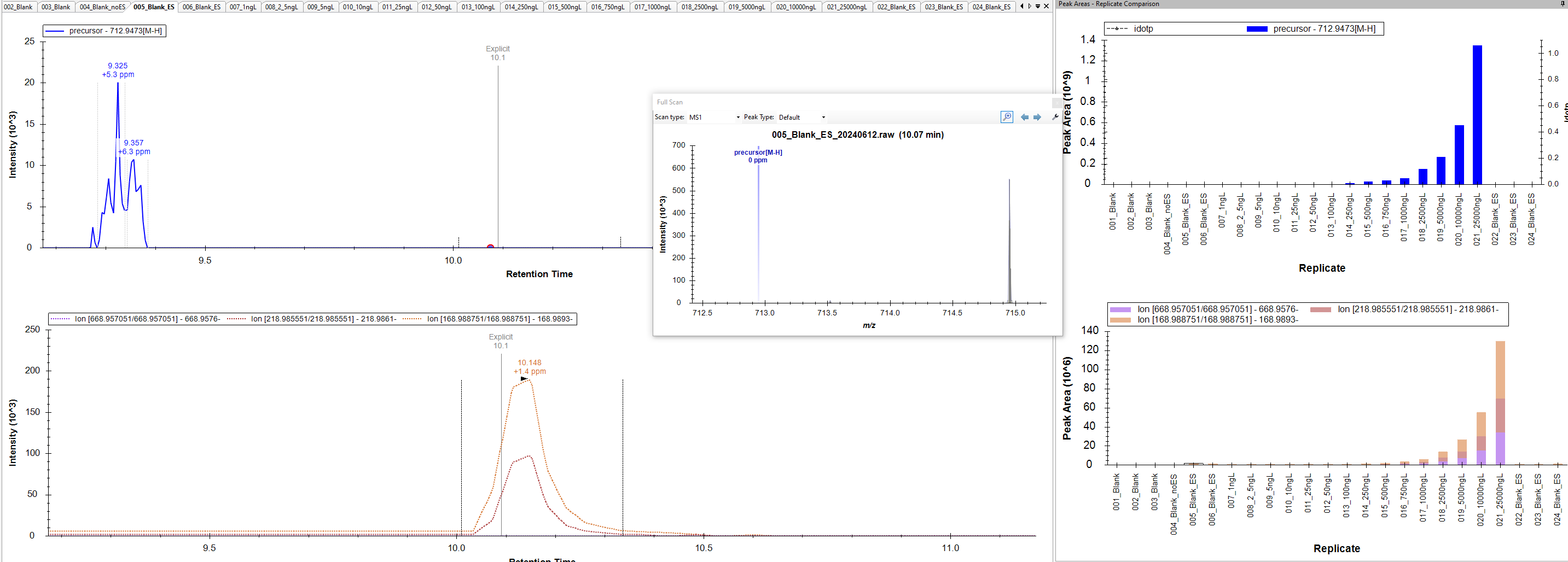 MS1-Spectrum-BlankSample.PNG
MS1-Spectrum-BlankSample.PNG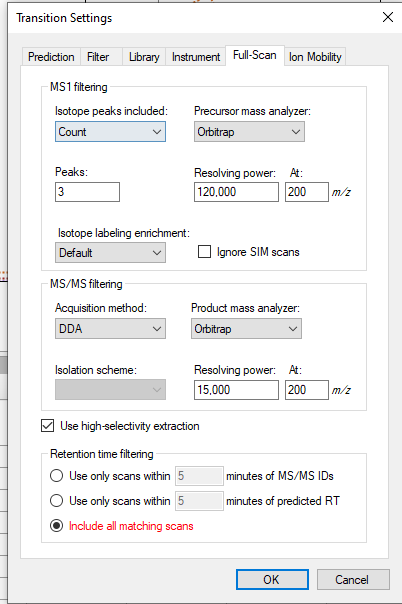 TransitionSettings.PNG
TransitionSettings.PNG