| Nick Shulman responded: |
2024-05-31 08:17 |
Can you send us your Skyline document and your .wiff and .wiff.scan files?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files.
Files which are less than 50MB can be attached to these support request.
If your files are larger than that you can package them up in another .zip file and upload them here:
https://skyline.ms/files.url
The error "sequence contains no elements" is nearly always caused by a programming error on our part.
After we see your Skyline document and .wiff and .wiff.scan files we will probably be able to figure out what is going wrong.
-- Nick |
| |
| george wang cal responded: |
2024-05-31 09:15 |
Thanks Nick. I've uploaded the .zip file there: "240531 george wang cal".
It is stdmix3 that is failing to import (included in the .zip). |
| |
| Nick Shulman responded: |
2024-05-31 09:45 |
Thanks for sending those files.
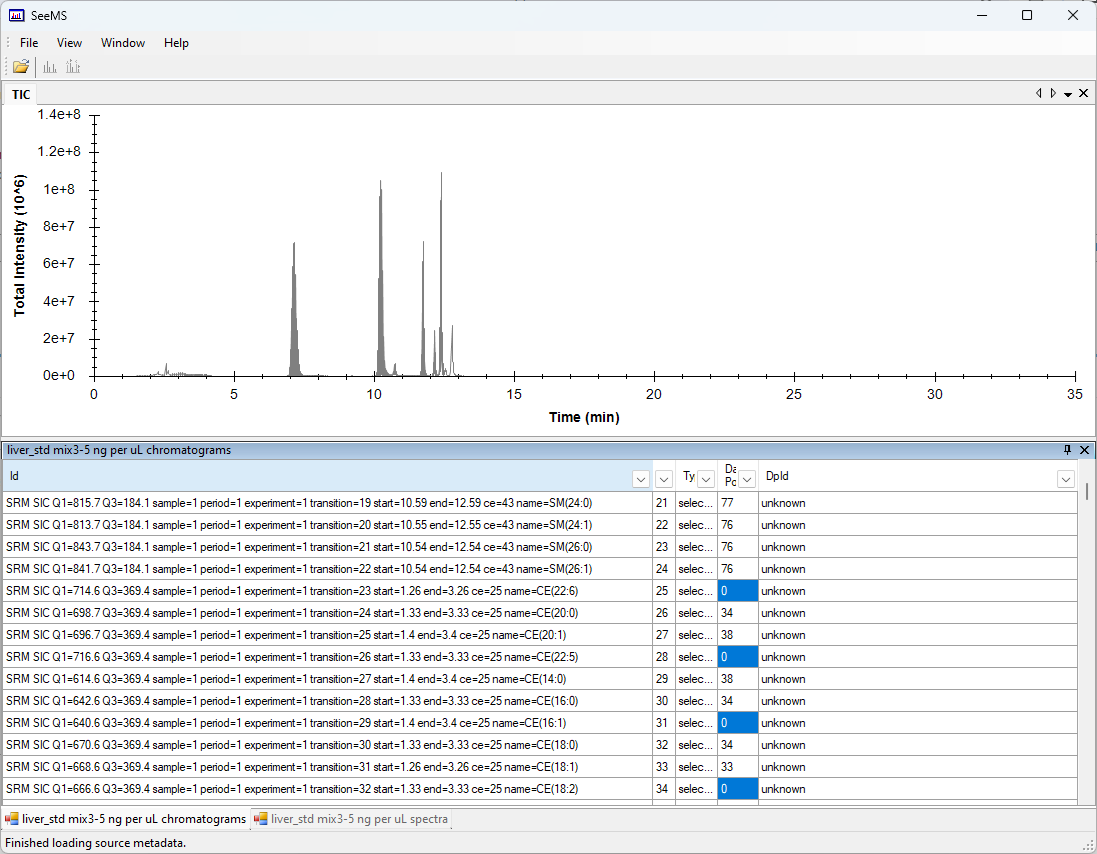
I looked at the file "liver_std mix3-5 ng per uL.wiff" using ProteoWizard SeeMS.exe.
There are several chromatograms in that file which have zero points in them.
I will fix Skyline so that it does a better job of handling this scenario.
Do you have any idea why some of those chromatograms have zero points in them?
-- Nick |
|
| |
| george wang cal responded: |
2024-05-31 10:54 |
Sounds good about the fix.
For those chromatograms it seems there is just no data there. I'm using the same acquisition method across the batch. Out of those 900 transitions, only 14 are for standards, and those are deuterated. Thus, the standard vials don't have any amount of the 886 remaining lipids, and should just have noise measured, while the samples don't contain those 14 deuterated transitions. It could randomly create a scenario where the standard vials have 0 points. What do you think? |
| |
| Nick Shulman responded: |
2024-05-31 11:41 |
If the mass spectrometer does not detect any signal, then what we would expect to find is that the chromatogram has some intensity values in it, but the value of each of those intensities is zero.
However, what we see is that there are no intensity values at all, zero or otherwise.
This does look very suspicious because the chromatograms with no values do have a definite start and end time (e.g. 1.26 to 3.26 minutes), so the mass spectrometer thought it was collecting data across a real time range.
I will ask my coworkers and find out if Skyline is interpreting this data incorrectly.
-- Nick |
| |
| george wang cal responded: |
2024-05-31 13:05 |
Yes, the mass spectrometer is definitely collecting data across that range. The 714.6-> 369.4 has a few data points in the "stdmix4" sample, intensity is very low of course ~300 counts.
Just thought of another possible explanation: this compound elutes at 2.4 min, and there are many transitions in this retention time range. There are about 400 total spanning both modes, meaning there will be only 6-7 points acquired baseline to baseline, or only 3 per peak. I've counted the # of points in my samples. That, combined with the zero abundance, could combine to cause this issue? |
| |
 SeeMSChromatograms.png
SeeMSChromatograms.png