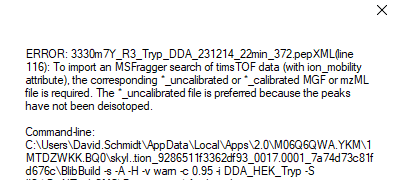
For that file "3330m7Y_R3_ChymoDDA_231214_22min_373.pepXML", BiblioSpec would like to find a file that might be called "3330m7Y_R3_ChymoDDA_231214_22min_373_uncalibrated.mgf" or "3330m7Y_R3_ChymoDDA_231214_22min_373.mzML" (or any of a long list of other possibilities).
It would only be looking for something with "_uncalibrated" in its name if it ended in ".mgf". For the ".mzML" extension BiblioSpec is not trying to insert the "_uncalibrated".
You have a different pep xml file in there "interact-3330m7Y_R3_ChymoDDA_231214_22min_373.pep.xml" which thinks it was produced from a file called "3330m7Y_R3_ChymoDDA_231214_22min_373_uncalibrated" so it's going to be looking for files called "3330m7Y_R3_ChymoDDA_231214_22min_373_uncalibrated_uncalibrated.mgf" or "3330m7Y_R3_ChymoDDA_231214_22min_373_uncalibrated.mzML".
You might be able to get this to work if you use "interact-3330m7Y_R3_ChymoDDA_231214_22min_373.pep.xml" instead.
Alternatively, you might be able to get this to work if you rename some of the files that you have to what BiblioSpec is expecting. However, if you rename things, sometimes you are able to successfully create a spectral library but, because the wrong file type was used, and there was ion mobility involved, the spectra that were included in the library are the wrong ones. You can tell that this has happened because when you go to "View > Spectral Libraries" the spectra that you are shown have very very few fragment ions that match the peptide that was supposedly detected.
If you send me all of the mzML and mgf files you have I might be able to give you more information about what to do.
-- Nick |
 Error_Library_Building.PNG
Error_Library_Building.PNG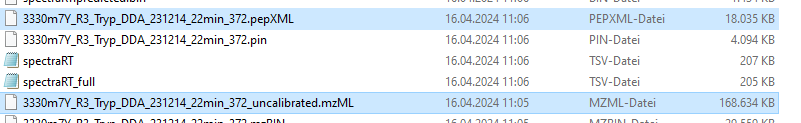 Files.PNG
Files.PNG