Hi Anne,
Can you explain why you would want different boundaries for light and heavy? After careful consideration and years of experience and feedback from researchers using Skyline, we decided to always integrate light and heavy peaks with the same boundaries. We believe this is the most correct peak integration you can possibly have, because with the majority of isotope labeled peptides we use, chromatography is identical for light and heavy. By integrating them differently, you are in fact introducing error into your measurements. If one or the other has interference that you cannot exclude by dropping a transition, then the most correct measurement you can possibly make is to integrate the time range without interference exactly the same for both peaks.
When you have isotope labeled reference peptides, the most important measure of abundance for your target peptides is the ratio between light and heavy, which is actually valid over any range you choose to integrate for your peak, as long as your range contains at least the start or end of your peak for proper background subtraction, and as long as you integrate exactly the same range for both light and heavy. Think of each point on the chromatograms Skyline displays as a sampling of that abundance. Assuming you have correct background subtraction, then your ratio at each point is a valid measure of peptide abundance, just as a single spectrum can be used in isobaric tagging methods like iTraq. Having the entire peak will give you the most precise measurement possible, but if you have matching reference peptides, you can get valid measurements without the entire peak.
Conversely, if use a different number of points or unbalanced sampling of your ratio, then you will not have a valid measure of abundance.
Only in cases where chromatography actually differs between your light and heavy, like deuterium labeling, would you want different integration boundaries. To get that, you have to specify that you are using this type of labeling, when you define your isotopic label modification and chose something other than "Matching" in the "Relative retention time" dropdown list (e.g. Overlapping, Preceding or Unknown). I do not recommend these for isotopic labeling that is not expected to affect elution times.
We have also left one final loophole, though, I also do not recommend using it. It is that you can still always integrate any individual transition however you like without impacting others. You need to use View > Transitions > Single. If you use manual integration in this mode, which I hope very few people will, it is still possible to integrate a single transition differently from the rest. I would recommend, however, that you first consider excluding any transition where you feel this is necessary from your measurements, as otherwise, you are likely adding error.
Hope that helps clarify. Please feel free to explain in more detail and provide screenshots for the case where you want to do this.
--Brendan |
Thanks so much for your complete answer.
I fully agree with you regarding the retention times between light and heavy peptides. However, I have a very specific experiment where I know that retention time of heavy peptide is modified.
Your explanations were perfect, so thanks again.
Anne |
Hello,
I try to develop a new SRM method with heavy peptides measured with orbitrap to have the retention time and the MS/MS of all the peptides and then export the SRM method.
I have the opposite problem. I would like to define the peak boundaries only with the heavy peptides (I did not saw the light peptides with the untargeted method). To export my SRM method i would like that the boundaries are only define by the heavy but when I export the method I saw that Skyline did a mean between the retention time of the heavy and the light. The light and heavy will have the same boundaries but the light also impact this boundaries despite the fact that I did not detect clearly the peak with the untargeted method (Orbitrap Q-exactive). When I move the boundaries in the heavy it moves it in the light but this is never exactly the same boundaries. Is it possible to fix this problem ?
Regards,
Nicolas Pierre |
Hello,
I wanted to follow up on this question. I would also like the capability to integrate the light and heavy separately (right now in order to do this, essentially I am renaming the light/heavy to show up as their own "molecule" (e.g., alanine_light and alanine_heavy) rather than being integrated under the same "molecule name" (e.g., alanine).
Our reasoning for doing this is we have a consistent offset with our light peak due to the D5 label. Typically we can set a wider integration window allowing both peaks to fall within the integration range, however, I worry about instances where there may be an overlapping light peak in that window that I would rather exclude by only changing the bounds of my heavy integration.
I've attached an example image of the data for reference.
Thank you,
Brianna M. Garcia |
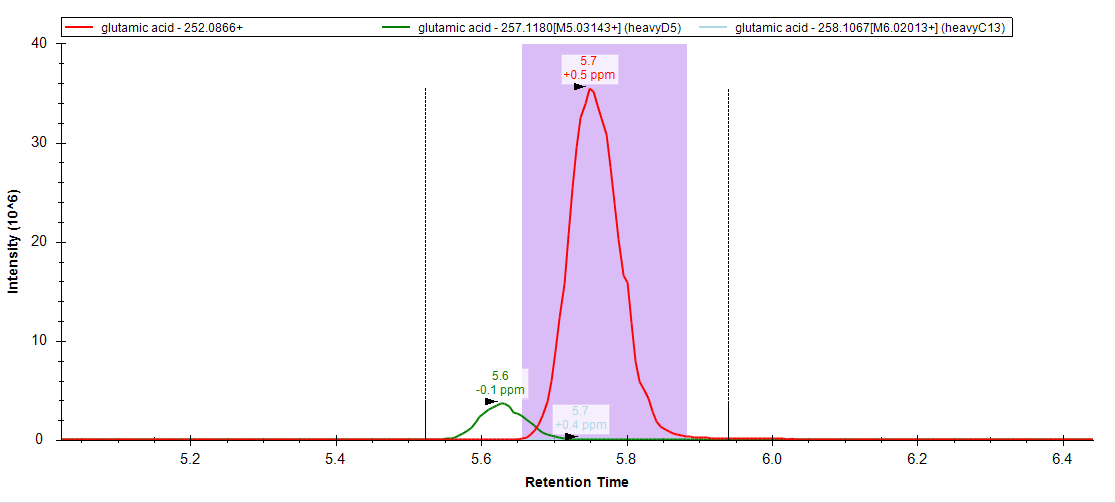 glutamicacid_Skyline_example.PNG
glutamicacid_Skyline_example.PNG