| a-ions from fully SIL labelled amino acids | jfoe | 2024-02-14 04:35 | |||||||||||||||||||||||||||||||
Dear Skyline-Team, I have a workflow where I create spectral libraries from Skyline. I recently noticed that I am missing some a-ions from my SIL-labelled peptides, if the fragmentation is next to the SIL label. The reason is, that while the common b- and y- ions fragment neatly at the peptide bonds, the a-ion loses a C-atom that belongs to one of the adjacent amino acids. I am including a screenshot that hopefully shows you why this causes me to lose signal. In that specific case there is actually some peak at the wrong a6+, too, but I am missing the main, correct a6+ peak. In effect, this is quite bad because the spectral library will end up with a bad dotp when detecting the natural non-labelled peptide because it expects only a very weak a6+ ion. Let me know if anything is unclear. Best wishes, |
|||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||
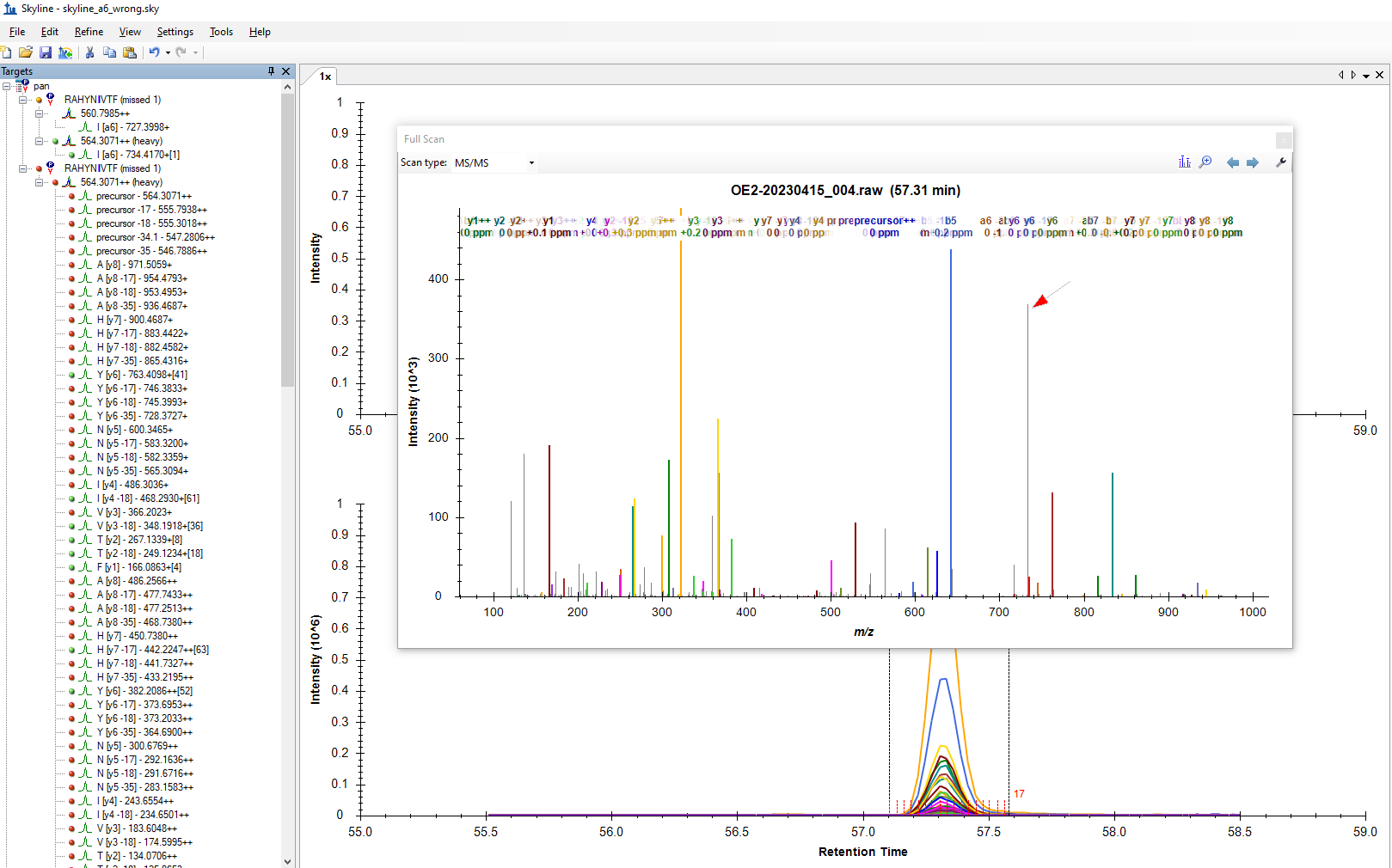 skyline_a6_wrong.png
skyline_a6_wrong.png skyline_a6_wrong_I_label_mod.png
skyline_a6_wrong_I_label_mod.png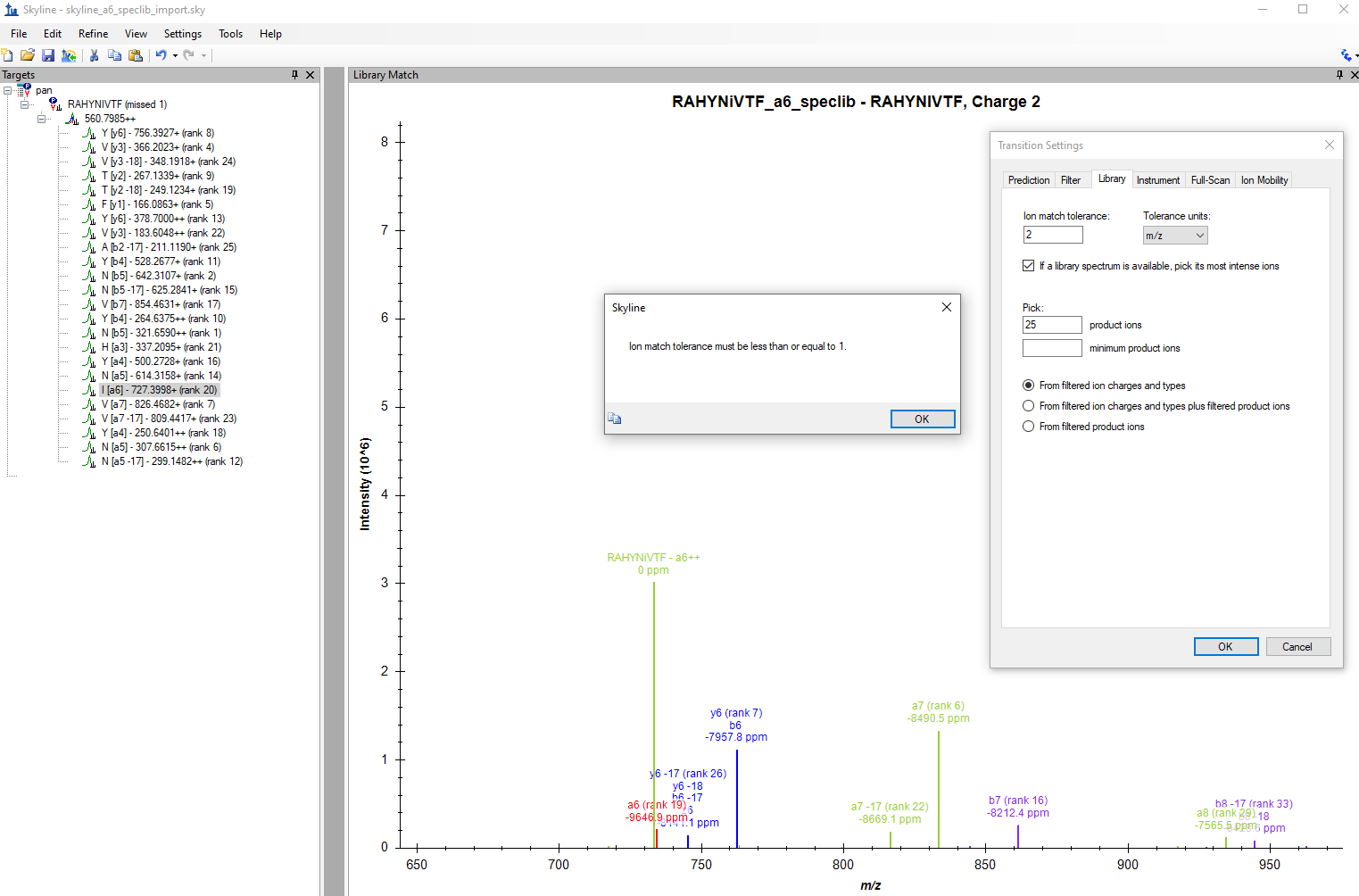 a6_special_ion_speclib.png
a6_special_ion_speclib.png