Hi Nick,
Thank for the link. This is the same question I asked. I'm processing lipid data for group comparison. Manually, I can export the raw data (area peak intensity) and calculate adjusted p-value. To calculate p-value, area peak intensity is generally transformed to log2 scale. The reason for log2 transformation is well-explained by a review paper (Analytical and Bioanalytical Chemistry,
https://doi.org/10.1007/s00216-023-04991-2).I copy a few sentences here: "One common assumption in univariate hypothesis tests is the normality of abundances. In lipidomic data, a strong right skew
in the raw abundances is often observed due to the presence of a few lipids with exceptionally high concentrations, so it is standard practice to apply data transformations in an attempt to obtain normality. Specifically, raw lipid abundances are often log transformed, and/or normalized, using values such as the total ion current (TIC), median abundance value, or others." Attached screenshot picture (log2.png) shows an example of t-test using area peak intensity and log2 scale. The log2 transformation yields better p-value. Boxplots were also shown.
Thus, I would like to have an option in group comparison to have data in log2 scale instead of area of peak intensity. Otherwise, I will have to export the abundance data (area of peak intensity) and calculate adjusted p-value and fold change manually for each lipid.
Honghui
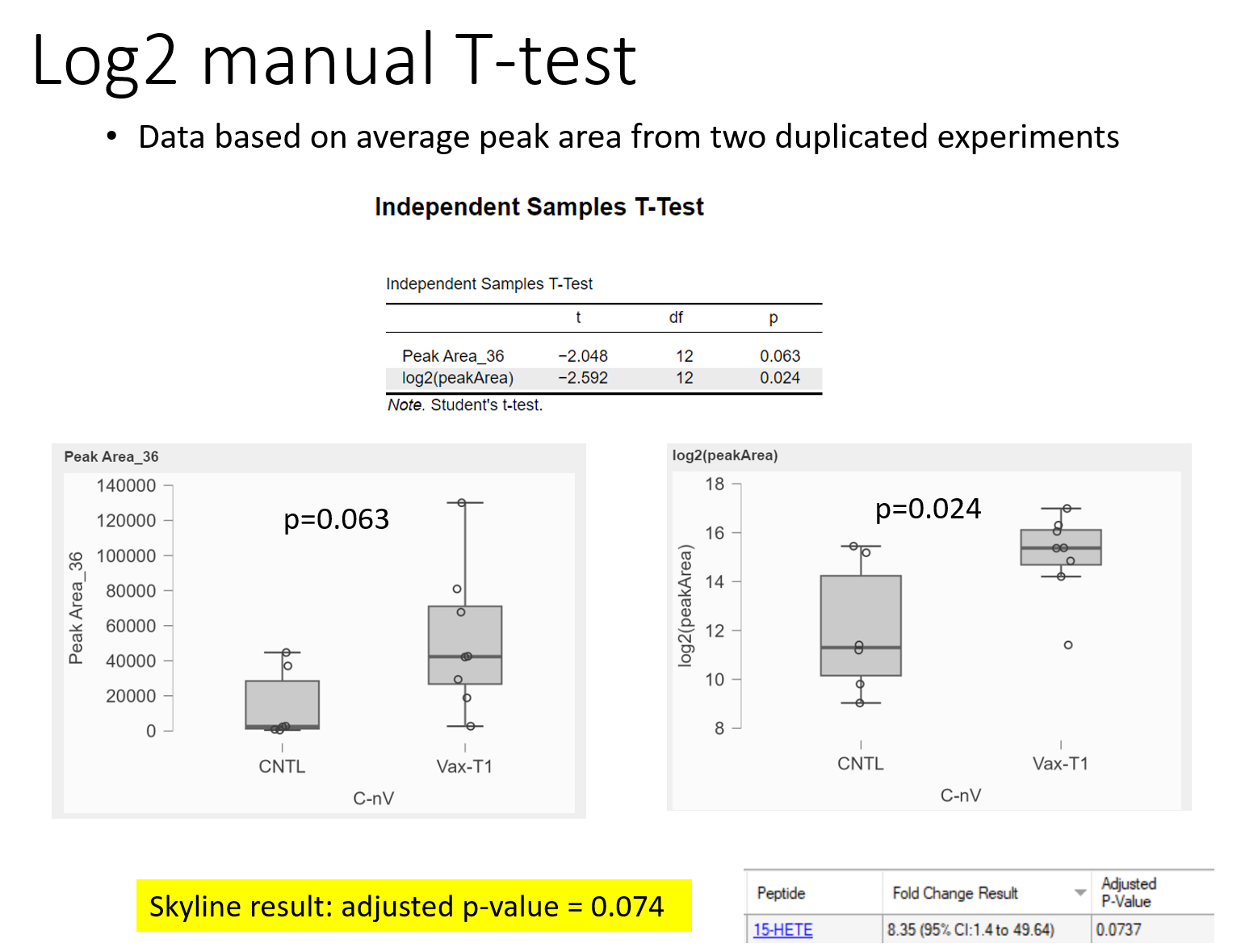 log2.png
log2.png