Hi,
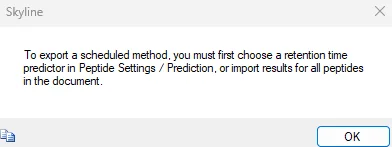
I am trying to build and export a scheduled PRM method (in a form of isolation list) from Skyline. When I'm done with the process I'm unable to export the method since "results for all peptides are not imported" (see "scrn1.jpg"). And no, I do not want to use RT predictors. It seems I am doing something totally wrong - but what?
I of course checked Skyline tutorials, but there is close to no mention of how to achieve my goal. Basicly, I've tried to build a spectral library using DDA data and PD results that I already have. I checked if the spectral library I'm using contains RT data - it does (see "scrn2.jpg"). It seems that Skyline wants results to let me export a method, but as I understand it, results should not be required since I've build a spectral library from DDA data already and that information is used to select candidate peptides for PRM analysis. Importing results onto candidates from this spectral library also results in missing/nonsatisfactory data (yellow and red dots next to peptides/transitions). Anyone has any idea of what to do to get start / stop times into the exported list automaticlly?
In the end I would like to use DDA data from samples to select candidates and build a scheduled PRM method - how can I achieve this?
Thanks!
|
| |
| Nick Shulman responded: |
2023-10-31 09:45 |
If your Skyline document contains chromatograms and a peak has been chosen for every peptide, then I think you just need to go to:
Settings > Peptide Settings > Prediction
and make sure that the "Use measured retention times when present" checkbox is checked.
If you do not have any chromatograms in your document, then you might be able to create an iRT predictor.
If you have chromatograms in your document, then Skyline has the ability to automatically choose a set of iRT standards from the peptides that you have, and create a whole iRT predictor using that.
I don't think there is a way to get Skyline to automatically choose iRT standards if all you have is a spectral library.
You might be able to go to:
Settings > Peptide Settings > Prediction
and push the calculator button and choose "Add..."
and then choose "CiRT (iRT C18)" from the "iRT standards" dropdown in the "Edit iRT Calculator" dialog.
The CiRT peptides are a set of peptides which are likely to be found in my different types of organisms, so it's likely that your spectral library already contains enough of them so that Skyline will be able to do a linear regression and assign iRT scores to all of the other peptides in your spectral library.
After you have chosen a set of standards, you can use the "Add" button at the bottom of the "Edit iRT Calculator" dialog and choose "Add Spectral Library" from the menu that appears when you push that Add button.
If that works, Skyline will display the "Add iRT Peptides" window which will tell you what slope and intercept Skyline used to convert the retention times in the spectral library into iRT scores.
Make sure you remember the numbers which are displayed in the "Equation" column in the grid in the "Add iRT Peptides" window.
After you have created your iRT predictor, I think you might also need to tell Skyline what slope and intercept to use to convert these iRT scores to actual retention times. You would do that by going to:
Settings > Peptide Settings > Prediction
and then in the "Retention time predictor" dropdown choose "Edit Current" and then uncheck the box which says "Auto-calculate regression" and fill in the values for the Slope and Intercept that you got from the Equation column in the Add iRT Peptides window.
It might be that there is an easier way to do what you are asking, in which case I hope someone will be able to add to this answer here.
-- Nick |
| |
| Daniel Fochtman responded: |
2023-10-31 11:54 |
Hey, thanks for the reply!
The "Settings > Peptide Settings > Prediction > Use measured retention times when present" has been checked and it does not allow to export the scheduled PRM method. This is exactly what I would like to use.
I do have chromatograms from DDA runs of my samples, so the RT is present and known a priori (that is shown in "scrn2.jpg" from the spectral library view). I would like to avoid using iRT prediction, as I mentioned in the original post, since that is just a predictor and I have actual RT available - thus, I would just like to use "Use measured retention times when present" option.
Again, after building the spectral library from DDA data Skyline choses peptides and transitions for proteins that I have already identified in that DDA data as targets for scheduled PRM. Yet, after importing this DDA data as a result with chromatograms attached, Skyline is unable to match peaks to peptides. How is this possible if this was used to build the library and chose these peptides in the first place? Why am I not able to build the method with RT times present?
As in the original post - in the end I would like to use DDA data from samples to select candidates and build a scheduled PRM method.
Thanks for the reply anyways though! |
| |
| Nick Shulman responded: |
2023-10-31 12:35 |
If you send us your Skyline document we might be able to figure out what you should do.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB you can attach it to this support request. You can upload larger files here:
https://skyline.ms/files.url
-- Nick |
| |
| Daniel Fochtman responded: |
2023-11-03 03:36 |
Hi,
I've sent over the Skyline document, spectral library and some examples of DDA raw files. All of this data can be now found as "PRM_export.zip" in the Skyline file sharing tab.
Thanks for the help! |
| |
| Nick Shulman responded: |
2023-11-03 04:36 |
Thank you for uploading that Skyline document and raw files.
The Skyline document in there did not have any chromatogram, but I used the "File > Import > Results" menu item to extract chromatograms from the raw files that you had included.
Unfortunately, even if your document has chromatograms, you will not be able to export a scheduled method unless every peptide in the document has a chosen peak in at least one of your replicates.
The error message that you are getting unfortunately does not tell you which peptide does not have any chosen peaks.
I can tell you that one of the peptides which has no peaks was VHVASVNNFPTAAGLASSAAGYACLAYTLAR.
The chromatograms for that peptide are completely flat, so Skyline was unable to choose a peak.
You can use the Document Grid to find the peptides that do not have any chosen peaks.
In the "Peptides" report, if you filter for all of the rows where "Average Measured Retention Time" is blank, you can see the peptides which have no chosen peaks in any replicate. In my document, there were 9 peptides with missing peaks, and after I deleted those peptides from the document I was able to export a scheduled method.
If you would like to learn more about the Document Grid you can look at the Custom Reports tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_custom_reports
-- Nick |
| |
| Daniel Fochtman responded: |
2023-11-03 05:06 |
Ok, thank you. This information is crucial and very useful. This will allow me to build a scheduled PRM method in the end, but can you advise me on some details so that I fully understand what is going on.
How would Skyline choose peptides as “best” for my proteins if the spectral library was build using DDA files, yet these peptides have no RT times attached to them in the "Peptides" report? What would be a better practice – building own library from DDA, importing that DDA as results and then deleting missing data OR using an already premade library (like NIST), importing DDA data then deleting missing results?
Thanks for this help! |
| |
| Nick Shulman responded: |
2023-11-07 10:22 |
I do not understand your new question.
What do you mean by "Skyline choose peptides as best"?
It sounds like you might be asking how Skyline would choose, for a given protein sequence, which peptides to add to the document.
Skyline uses the settings on the "Digestion", "Filter" and "Library" tabs of the "Settings > Peptide Settings" dialog to decide which peptides to give you. Skyline gives you all of the peptides which match those criteria: Skyline never compares two peptides that match the criteria in order to give you the "better" one.
I might be misunderstanding your question because you seem to also be asking about retention times.
I am not sure I understand the other part of your question.
You might want to skip the DDA part of your process because there are some peptides which can be detected via PRM, but which cannot be found by DDA because the MS1 signal is not strong enough for the mass spectrometer to notice them and select them for fragmentation.
Instead, what you could do is create a Skyline document with no spectral library, and all all tryptic peptides to the document.
After you have done that, you can go to:
Settings > Peptide Settings > Library
and push the "Build" button and choose the "Prosit" for the "Data source" in the Build Library dialog.
It used to be that you needed to do a DDA experiment so you might have some idea which transitions to look for, but, now, you can usually just get away with having Prosit predict which transitions you should look for.
-- Nick |
| |
|
|
 scrn1.jpg
scrn1.jpg scrn2.jpg
scrn2.jpg