Can you send us your Skyline document and one or more of your .raw files?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file and/or your .raw file are less than 50MB you can attach them to this support request.
You can upload larger files here:
https://skyline.ms/files.url
When your MS2 transitions are missing their chromatograms like they are in your screenshot, it is because Skyline did not find any MS2 spectra which matched the precursor in your Skyline document.
There are a couple of reasons that changing the Acquisition Method to "SureQuant" might have caused this to happen:
1. When you change the Acquisition Method to "SureQuant", Skyline also changes the "Method match tolerance" at "Settings > Transition Settings > Instrument" to "0.007". It is possible that this value is too small of a number and, for this reason, none of the MS2 spectra in your .raw file match the m/z of the precursor in your document.
2. When "Triggered chromatogram extraction" is checked at "Settings > Transition Settings > Instrument", Skyline pays attention to the "Scan Description" values on the spectra. If the scan description starts with "SQ_" then the scan description has to match a very specific format in order for Skyline to use it in a particular precursor's chromatogram. You can get more information about that here:
https://skyline.ms/announcements/home/support/thread.view?rowId=49364
After I see your Skyline document and raw file I will probably be able to tell you what is going wrong.
-- Nick
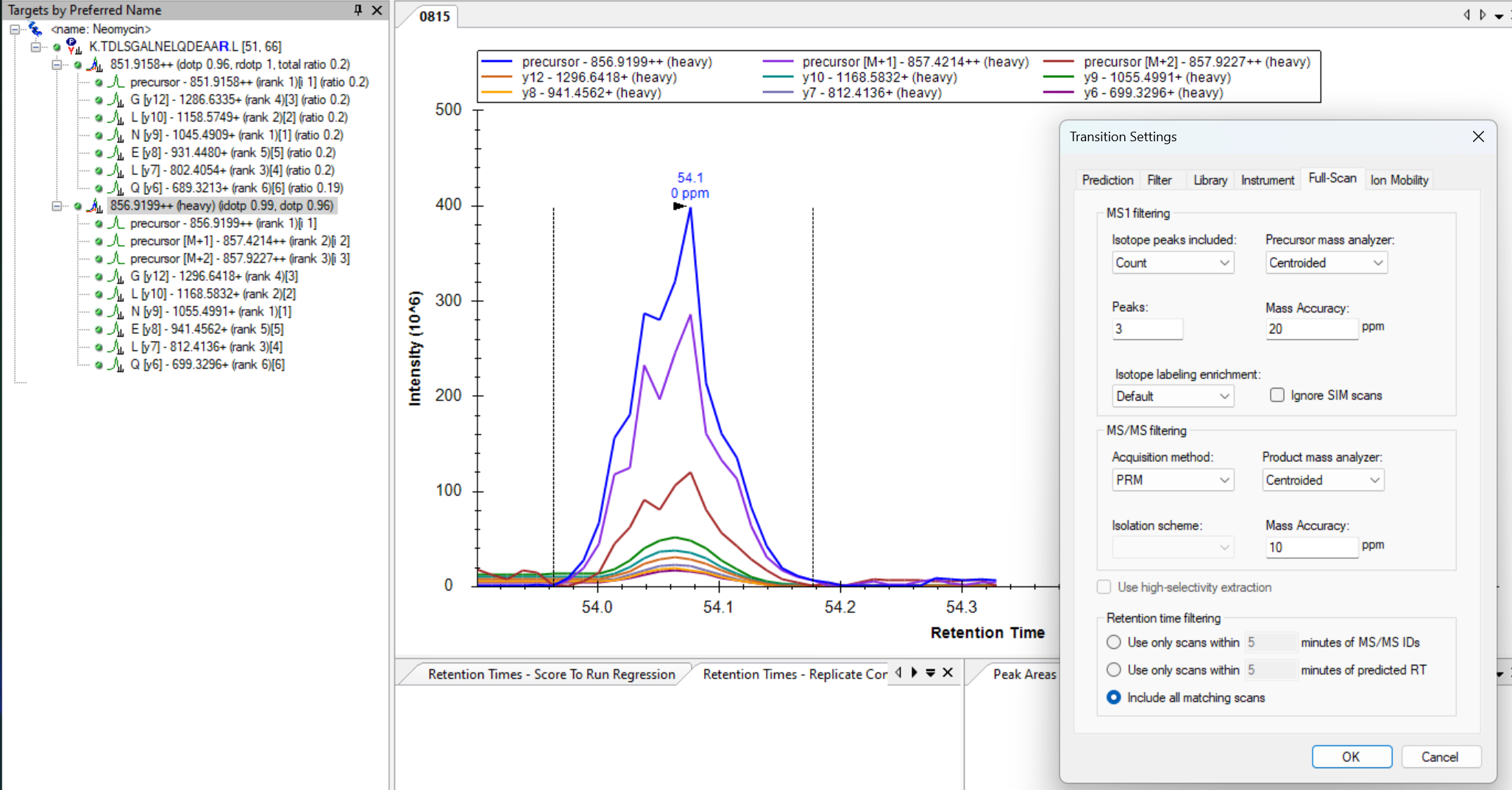 PRM.png
PRM.png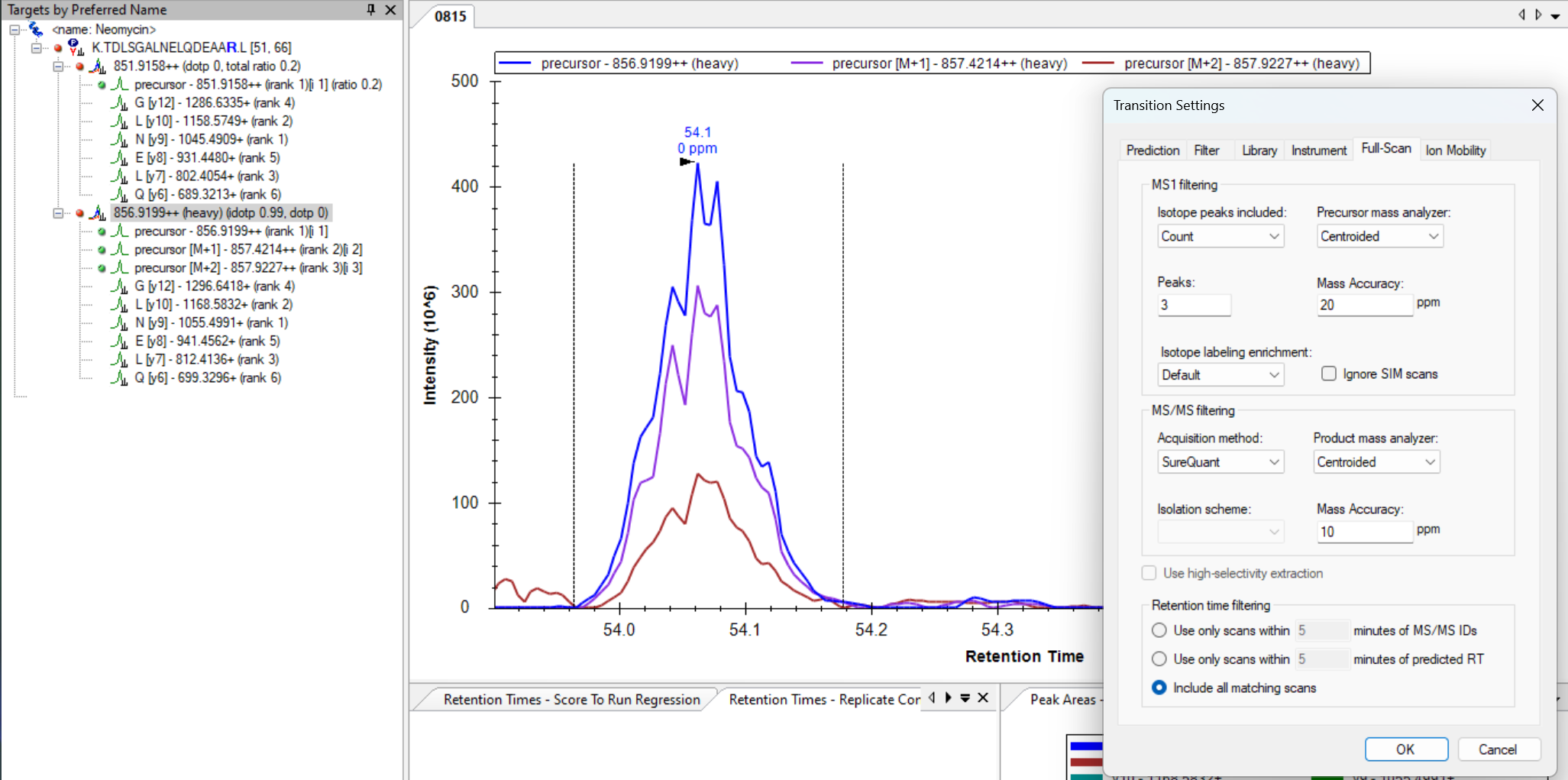 SQ.png
SQ.png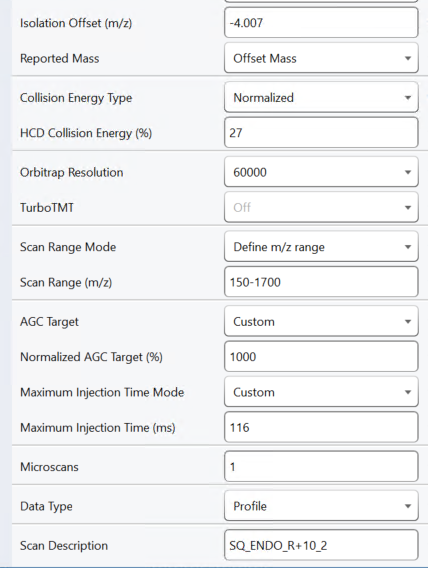 SD.png
SD.png