| Nick Shulman responded: |
2023-05-24 19:08 |
Can you send us your files?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
You should send us that .zip file as well as some of your .wiff and .wiff.scan files.
If those files happen to be less than 50MB you can attach them to this support request but they will probably be too big so you should put everything in a big zip file and upload them here:
https://skyline.ms/files.url
I am not sure I understand the behavior you are describing. It might be helpful if you could post a couple of screenshots.
-- Nick |
| |
| becker78 responded: |
2023-05-24 19:44 |
Hi Nick,
I uploaded the files.
The skyline file called 230524_PeptidesList_ParedToOptwDMSO is what I've been working with today.
Looking at the 4th peptide on the list DITSD you can see the DP optimization looks strange. If you look by each transition the y10 is missing half the data.
Then go to the oxidized methionine version of "STLD" the y9 +2 fragment is also missing about half the data.
Similar behavior for GNP the y8 and y10 are both partial.
Then look at LAN and the MGQ with and with out oxidized methionine. They each only show the skyline predicted DP and none of the other optimization data.
So then I created a fresh skyline file called 230524_DMSO_FreshFile_DPOpt and reimported the data.
The STL, DIT and GNP are still broken. But LAN, MGQ, and MoxGQ peptides are somewhat better but still missing some of the data.
LAN is missing b4
MoxGQ missing y8 transitions
MGQ also missing y8 transitions
GNP is also broken
I have some data from last week where it was doing a similar thing with collision energy data too if sending more info along would help you.
Thanks so much for looking at this issue! |
| |
| Nick Shulman responded: |
2023-05-24 21:15 |
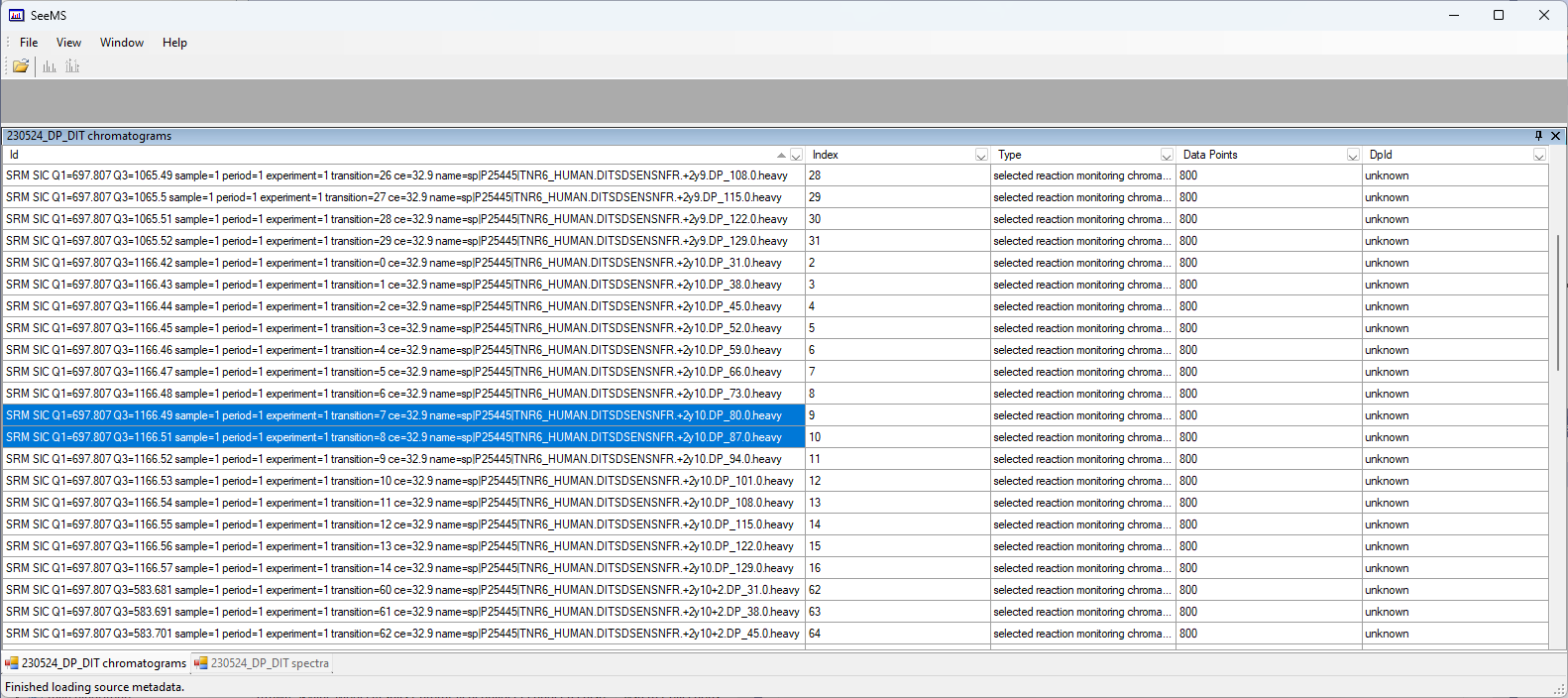
When I look at the wiff file "230524_DP_DIT.wiff", it seems to be missing some chromatograms.
I used the program "SeeMS.exe" which is part of ProteoWizard to look at that wiff file.
When Skyline is exporting a method for optimization, Skyline tells the mass spectrometer to collect data for a series of transitions with the same Q1 value, but where the Q3 value is a series of numbers centered around the actual product m/z but which differ by 0.01 units.
So, if you are collecting data for 7 optimization steps for the precursor m/z 697.8065 and the product m/z 1166.4948, then the mass spectrometer would be told to collect data for Q3 values ranging from 1166.4248 to 1166.5648.
When I use SeeMS.exe to look at the chromatograms that are in the wiff file, the chromatogram names have all been rounded off to two decimal places, which is probably to be expected, and so I see chromatograms going from 1166.42 to 1166.56, except that 1166.50 is missing.
Anyway, the missing chromatogram at 1166.50 causes Skyline to ignoring all of the higher optimization steps because Skyline does not have the concept of skipping over an optimization step.
Can you figure out why the chromatogram Q1=697.807 Q3=1166.50 is missing from that wiff file?
Did Skyline export the wrong transition list or did something else go wrong?
-- Nick |
|
| |
| Nick Shulman responded: |
2023-05-24 21:31 |
Actually, now that I look at this closer, I see that there really are 15 chromatograms there, which is the correct number of chromatograms for 7 optimization steps. The problem is just that there is an unexplained gap at 1166.50, as if something decided at that point to switch between rounding down to rounding up.
I do not know very much about method export or wiff files so I do not have a good idea about what might be going wrong. I am surprised that the Q3 values that we see in the chromatogram IDs only have 2 digits of precision. I was under the impression that they usually had at least 3 digits.
-- Nick |
| |
| becker78 responded: |
2023-05-25 13:16 |
Hi Nick,
I've been using skyline to export transition lists and then importing the .csv files into analyst rather than having skyline export a method file.
I opened a pair of my exported transition lists and .csv files. Analyst does seem to be truncating the number to 3 decimal points for the precursor mass but the fragment is in 3 decimals for everything.
I'm attaching an excel file with a tab that was the .csv that I exported from skyline.
Then I've copied those MRMs back out of the analyst method file I imported it into.
I don't see why, from what I can see, it would cause skyline to fail to import once it's done. The numbers look like they're matching.
-Jess |
|
| |
| becker78 responded: |
2023-05-25 13:36 |
Also I'm not sure it's all just rounding because if you look at the LAN peptide Q1 = 631.3657 analyst has collected all the MRMs. When I imported it into the skyline document I was working with 230524_PeptidesList_ParedToOptwDMSO it only shows one MRM imported and none of the optimization data.
When I copied and pasted that peptide into a new skyline file and reimported (230524_DMSO_FreshFile_DPOpt) then I can see most of the data but the b4 fragment Q3= 412.2554 is incorrect like some of the others.
I'm attaching a those exports for you to look at as well.
-Jess |
|
| |
| Nick Shulman responded: |
2023-05-25 15:06 |
With the chromatograms for the "LAN" peptide, the problem is that there are a couple of chromatograms which have a 6 in the third decimal place instead of a 5.
In order for Skyline to recognize a series of chromatograms as belonging to set of optimization steps, the difference between the Q3 values needs to be between 0.009 and 0.011.
The difference between 412.205 and 412.216 is ostensibly 0.011, except that computers do not represent decimal numbers exactly so I cannot say for certain whether Skyline thinks that (412.216 - 412.205) is greater or less than 0.011.
I guess to be absolutely sure that rounding errors are not going to cause chromatograms to disappear, it is necessary to make sure that the values have been specified to 4 decimal places.
Note that the behavior that you are seeing in Skyline-daily is a little different than it was in Skyline 22.2.
In Skyline 22.2 there were many cases where Skyline would keep counting past a gap in the Q3 values so long as the difference between the Q3 value that Skyline was looking at and the product m/z of the transition was less than the "Method match tolerance m/z" specified at "Settings > Transition Settings > Instrument".
-- Nick |
|
| |
 SeeMS_Screenshot.png
SeeMS_Screenshot.png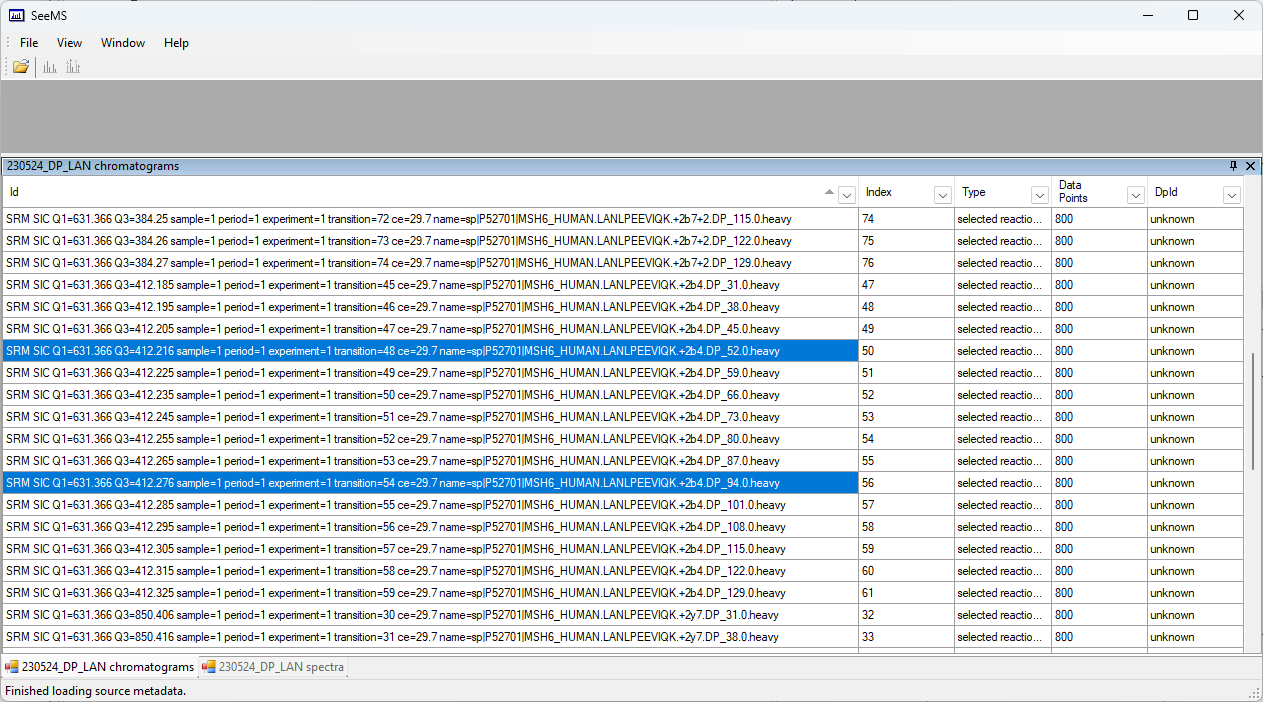 LAN_chromatograms.png
LAN_chromatograms.png