It sounds like you might have tried to attach your .sky.zip file to a support request, but nothing actually got attached.
This often seems to happen if the file you are attaching is larger than the 50MB limit.
You can upload larger files here:
https://skyline.ms/files.url
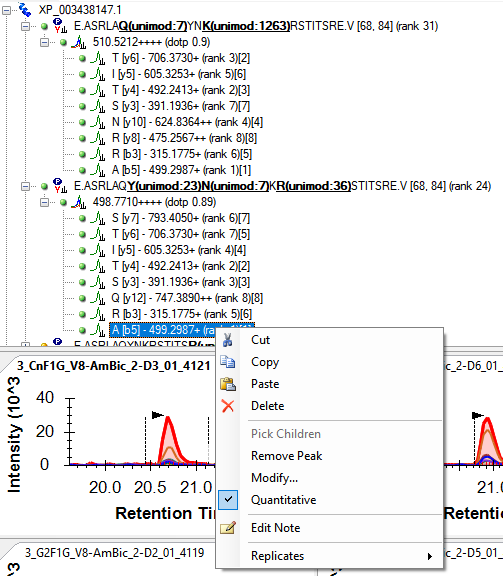
Usually, people decide which transitions have interference by having multiple technical replicates, rather than trying to predict which transitions are going to have the same retention time and precursor window as some other thing in the sample. The transitions with interference are likely to have larger CVs across the replicates, because the thing that is interfering is likely to vary more across the replicates in terms of how much it interferes with the measured signal.
I am supposed to implement some features in a future version of Skyline which will help you figure out which transitions have interference like that, and either remove them from the document or mark them as non-quantitative. I am not sure what the best way to do this with the current version of Skyline is. There might be something useful on the "Results" tab of "Refine > Advanced", or there might be something useful with "View > Peak Areas > CV Histogram".
Some people use other tools to decide which are the most quantitative transitions. One popular tool which we are planning on copying some features from is EncyclopeDIA:
https://bitbucket.org/searleb/encyclopedia/wiki/Home
-- Nick
 Screenshot.png
Screenshot.png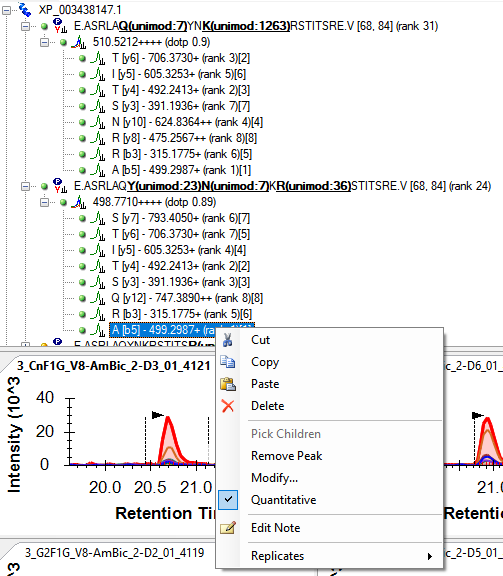 Screenshot.png
Screenshot.png