Hi,
I hope you're well
I'm having trouble training a mProphet reintegration model that can separate targets from decoys. I have followed the advanced peak picking tutorial, but I am uncertain if there is an error in my Skyline document settings (or my spectral library) that may be contributing to this.
I had set my peptide and transition parameters in Skyline and then imported a spectral library generated by protein pilot (for approx. 8000 human proteins including PepCalMix iRT peptides) and generated decoys peptides (same number as target peptides) using either the shuffle or reverse sequence method. After importing DIA data, I trained a mProphet reintegration model but it does not separate the target peaks from the decoys. Also, I want to integrate peaks with a q-value <0.01, but most of the peaks in the model have large q-values
I noticed that multiple features were greyed out (retention time difference, library intensity dot product, etc) - I deleted the unknown peptides to use these features, but that did not improve the model.
If I integrate peaks with the default method, I get approx 1500-2000 proteins at q-value <0.01. I can identify a similar number of proteins (q-val <0.01) in my samples using the same spectral library with other software. I'm aware that the default method works quite well, however as I have a large target list, I was hoping to use the mProphet model for more accurate peak picking. Any support to understand and rectify this situation would be much appreciated.
Kind regards,
Susannah
|
| |
| Nick Shulman responded: |
2022-08-17 22:50 |
If you send us your Skyline document, we might be able to figure out what is going wrong.
In Skyline, you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB you can attach it to this support request. You can upload larger files here:
https://skyline.ms/files.url
By the way, there is a new feature in Skyline-Daily: the Candidate Peaks Grid. You can get to it with the menu item "View > Other Grids > Candidate Peaks". The Candidate Peaks grid will show you all of the peaks that Skyline detected for the currently selected peptide in the current replicate, and you will be able to see the value that the peak got for all of the scores that make up the current peak scoring model. We hope that the Candidate Peaks grid will give users much more insight into why Skyline is making the peak picking decisions that it is.
-- Nick |
| |
| susannah hallal responded: |
2022-08-18 21:56 |
Thank you. Sure thing, I've uploaded the skyline document using the link
Susannah |
| |
| Nick Shulman responded: |
2022-08-18 22:19 |
I see that you uploaded the .sky, .sky.view and .skyd files.
I am also going to need the files "Human_SH_Prot.protdb", "iRT_PepCalMixSH22.irtdb", and "GBMSkinLibrary_iRT_2022.blib".
It's usually best to upload the .sky.zip file that gets created when you use the "File > Share" menu item. Skyline packages all of the necessary files together in a single .zip file. That file will probably be about 20GB in size, and it might take you a long time to upload it to our server, but our server has plenty of bandwidth and storage space.
-- Nick |
| |
| susannah hallal responded: |
2022-08-18 23:36 |
okay, I uploaded my document as a .zip file now so that it has all the necessary files |
| |
| Nick Shulman responded: |
2022-08-19 02:09 |
Thank you for sending that Skyline document.
I see that you are using Skyline version 20.2. The latest released version of Skyline is 21.2.
We are going to be releasing Skyline 22.2 in a few weeks. If you want to see which features are going to be in this next version of Skyline you can install Skyline-Daily from the Skyline website.
There probably aren't any new features of these newer version of Skyline that would fix your particular problems, but the newer versions of Skyline do consume less memory with large documents like yours, and that alone is probably a good reason to upgrade.
I see that you have about 7000 proteins and about 100,000 non-decoy peptides.
It is probably not possible to detect anywhere that many peptides in your samples, so, when you train your peak scoring model, you really would expect that bell curve of the decoy scores would very much overlap with the scores of your target peptides, and, if you are lucky, there will be a tiny hump to the right of those two bell curves which represent the detectable target peptides.
I see that your peptides all have the light precursor as well as the 13C labeled heavy precursor. Do your samples actually contain heavy peptides in addition to the light ones? I see that "Internal standard type" at "Settings > Peptide Settings > Modifications" is set to "none", which means that Skyline will be equally interested in looking for light and heavy transitions.
I see that many of your peptides (e.g. DFTGAITLLEFK) do not have any chromatograms. It looks like the reason for this is that these peptides have a iRT score whose value is so high that the predicted retention time of the peptide is beyond the end of the experiment run. Your Retention Time Filtering setting at "Settings > Transition Settings > Full Scan" tells Skyline to only extract chromatograms in a five minute interval around the predicted retention time.
The iRT score for the peptide DFTGAITLLEFK is 113.44. The highest iRT score for any of your iRT standard peptides is "SGGLLWQLVR" with an iRT score of 94.08. (You can see the iRT scores if you go to "Settings > Peptide Settings > Prediction" and then press the calculator button and choose "Edit Current").
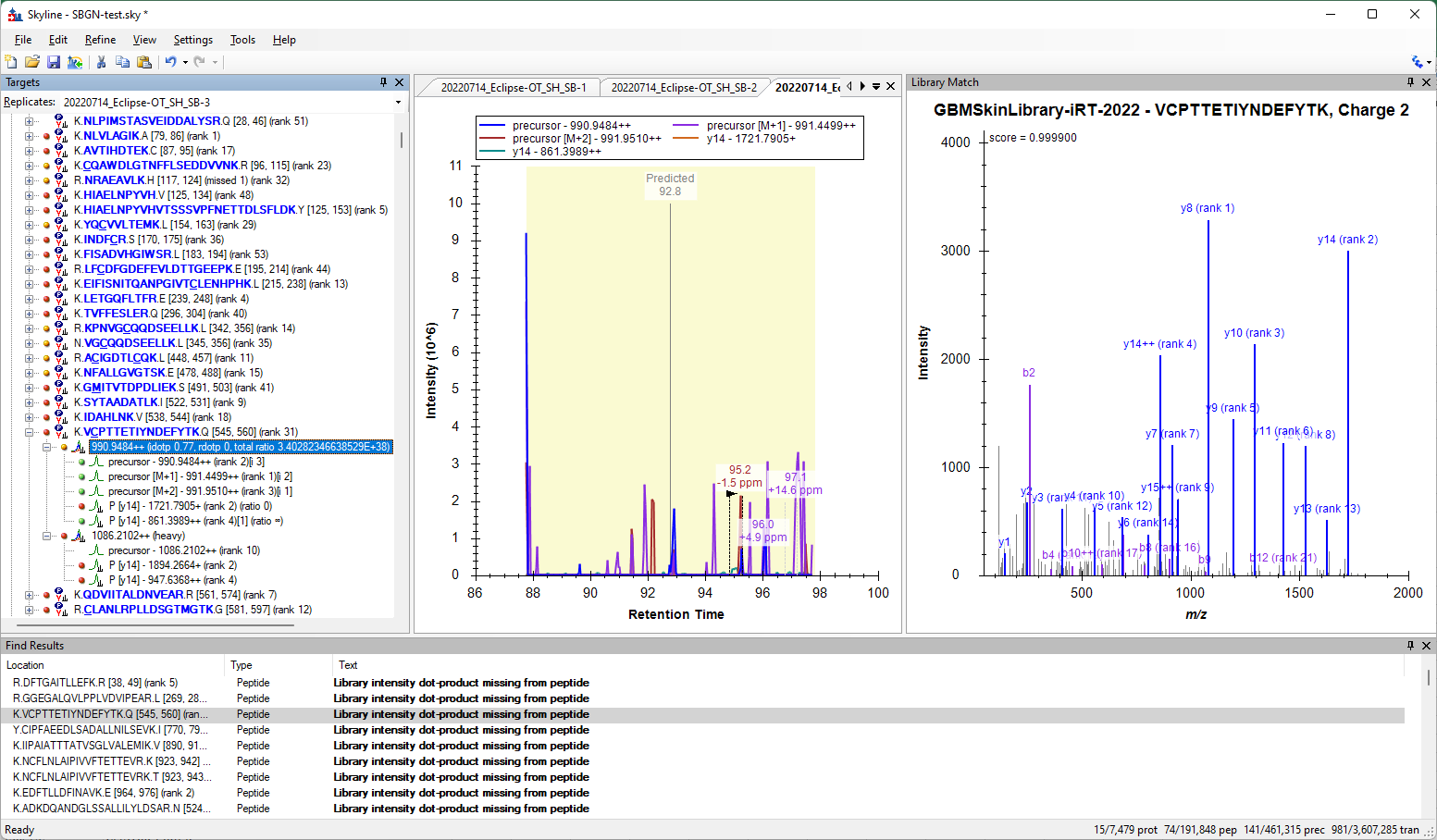
I also see that some of your peptides are missing the Library intensity dot product score. Most of those peptides are ones that are missing chromatograms, but there are some where the problem is that the peptide has only two transitions. Skyline requires at least 3 transitions in order to calculate a dot product score.
I am a little confused as to why the peptide in the attached picture only ended up with two transitions, even though the library spectrum has a ton of detected transitions. Usually something like this would be happening because your transition filter settings are preventing Skyline from choosing any of those transitions in the library spectrum, but I cannot see anything in your transition settings which could be causing this.
I will figure out why this is happening and get back to you.
-- Nick |
|
| |
| Nick Shulman responded: |
2022-08-19 03:46 |
Your document has both "Label: 13C15N" and "Label: 13C" checked at "Settings > Peptide Settings > Modifications".
You should uncheck one or the other of those, because Skyline is nonsensically trying to label the carbon atoms in the amino acid formulas twice.
I also figured out why Skyline is only giving you two transitions for the peptide "VCPTTETIYNDEFYTK". In the settings at "Settings > Transition Settings > Filter", the box "Use DIA precursor window for exclusion" is checked, which means that transitions will not be included if they have a m/z which is contained in the isolation window of the precursor. The peptide has both a light and a heavy precursor, so a transition will be excluded if it falls in either of those precursor's isolation window, which means that everything from 955 to 1650 is excluded.
Skyline only considers the top 5 transitions from the spectrum based on the "Pick 5 product ions" setting at "Settings > Transition Settings > Library". Of the top 5 ranking transitions in the spectrum, only y14+ (1721.7905) and y14++ (861.3989) are not in the range 955-1650.
I would have thought that Skyline would consider the sixth through eighth best transitions if three of the top five were being excluded for other reasons, but that is not how Skyline's transition picking works.
I think you will have fewer problems with this document if you remove the heavy precursors from your document. You can remove them by using the "Remove Label Type" option on the "Refine > Advanced" dialog. You could also remove the heavy precursors by unchecking both the "Label: 13C15N" and "Label: 13C" at "Settings > Peptide Settings > Modifications".
-- Nick |
| |
| susannah hallal responded: |
2022-08-21 17:05 |
Hi Nick,
Thanks so much for looking through my settings. I removed the heavy precursors and unchecked "Use DIA precursor window for exclusion" and its definitely improved the reintegration model
Susannah |
| |
|
|
 OnlyTwoTransitions.png
OnlyTwoTransitions.png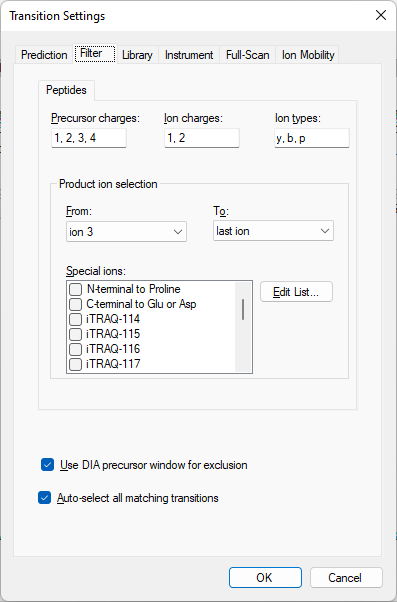 TransitionFilterSettings.png
TransitionFilterSettings.png