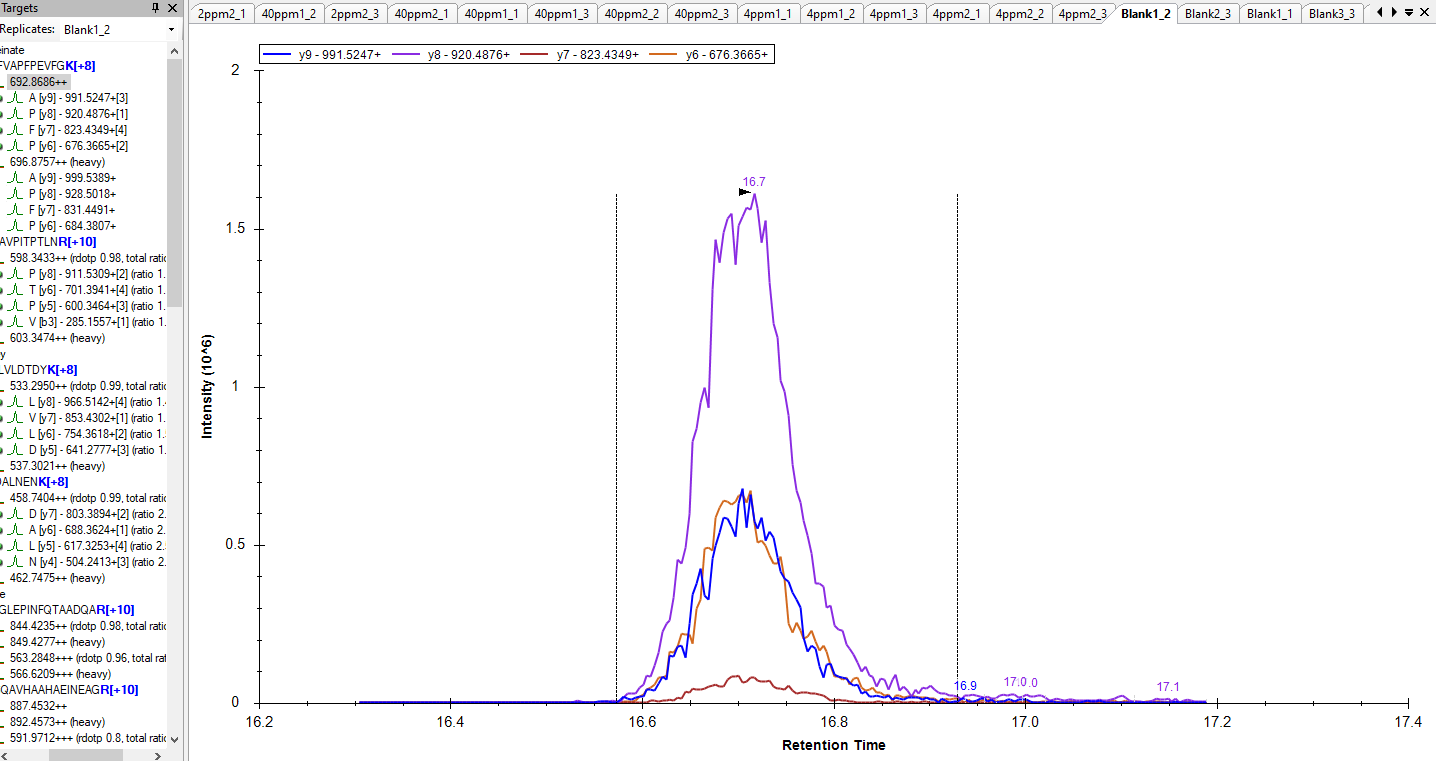
That probably means that Skyline could not find a chromatogram in your raw file whose Q1 and Q3 values matched the precursor m/z and product m/z of the transitions in your Skyline document.
I see in your screenshot that the precursor m/z is 696.8757 and the product m/z's are 999.5389, 928.5018, 831.4491 and 684.3807.
Was there a chromatogram in your raw file whose Q1 value was close to 696.8757 and whose Q3 value was one of those other numbers? The m/z values do not need to match exactly. The closeness of the match required is controlled by the "Method match tolerance m/z" setting at "Settings > Transition Settings > Instrument".
If you would like, you can send us your Skyline document and one of your raw files, and we can figure out what is going wrong.
In Skyline, you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file and your raw files are less than 50MB, you can attach them to this support request. You can upload larger files here:
https://skyline.ms/files.url
-- Nick
 Screenshot 2022-08-15 101957.png
Screenshot 2022-08-15 101957.png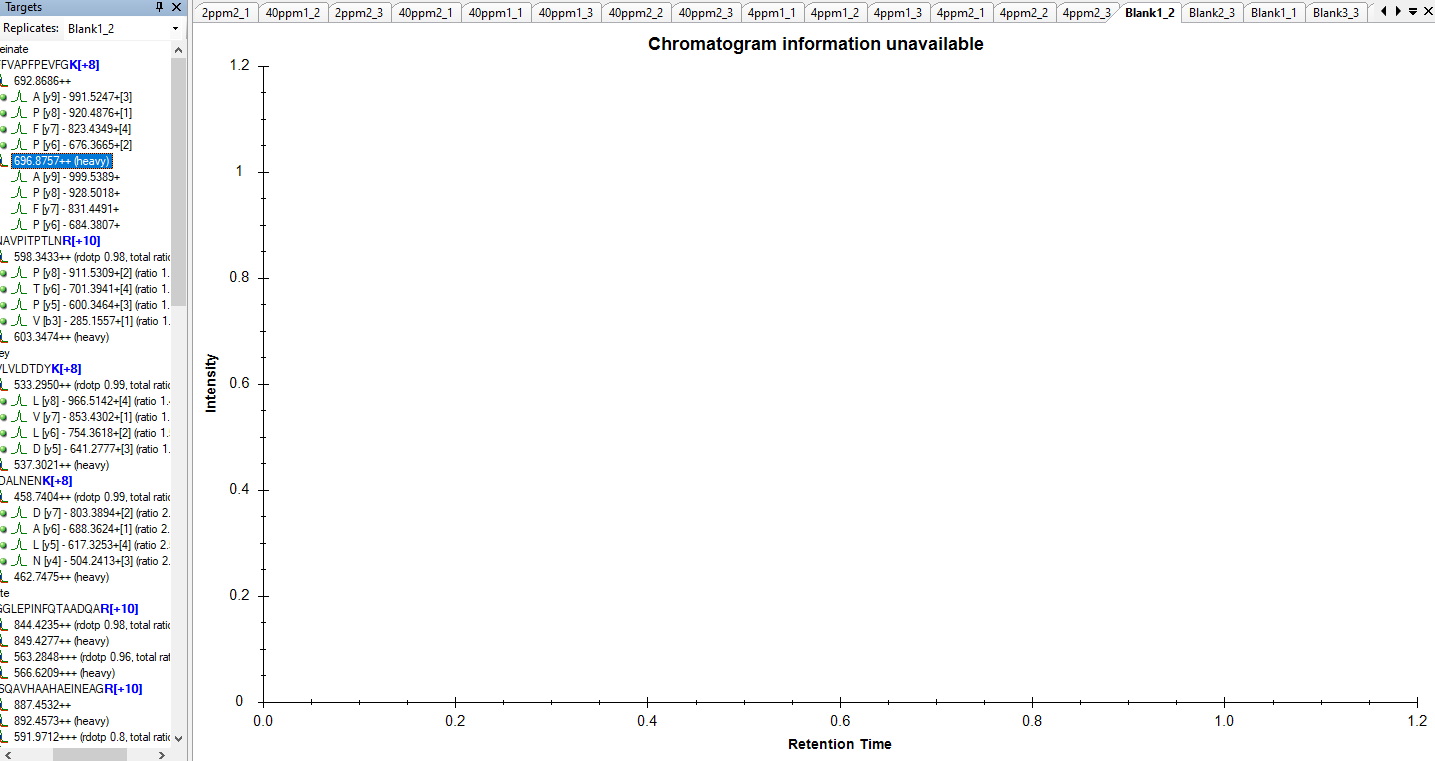 Screenshot 2022-08-15 102019.png
Screenshot 2022-08-15 102019.png