| Brian Pratt responded: |
2022-08-11 06:39 |
Hi Artu,
Changes to the ion mobility library won't have any effect until you re-extract chromatograms. You can use Manage Results for that.
Thanks for using the Skyline support board!
Brian Pratt
|
| |
| Brian Pratt responded: |
2022-08-11 07:38 |
P.S. If you already knew that, and this still isn't working, I'd love to see your Skyline document (use Skyline's File>Share menu item) and one of the data files in question.
Best regards,
Brian
|
| |
| Nick Shulman responded: |
2022-08-11 07:46 |
It sounds like Brian is correct that you just need to tell Skyline to extract chromatograms again by going to "Edit > Manage Results > Reimport" in order for your FAIMS changes to have any effect.
If you are still having trouble, you can send us your Skyline document and at least one of your .raw files.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file and your .raw files are less than 50MB you can attach them to this support request. You can upload larger files here:
https://skyline.ms/files.url
Sometimes with SureQuant methods, you can get spiky chromatograms if there is a heavy precursor in your Skyline document whose m/z is almost the same as the light precursor of some completely different peptide. The SureQuant method will have different settings for light and heavy peptides in terms of fill time and resolution, so, if a chromatogram is composed of different scans that were intended for different types of precursors, the chromatogram ends up spiky like what you have in your screenshots. In that particular case, the workaround that is available is to make it so that the mass spectrometer adds a "Scan Description" value to each of the spectra to tell Skyline which spectra belong to which peptide. The scan descriptions in this case would need to start with "SQ_" and after that they would have something indicating light or heavy and charge state. There is more of a description of what the scan description should look like here:
https://skyline.ms/announcements/home/support/thread.view?rowId=49364
But, this "Scan Description" thing would never be necessary in cases where Skyline is not doing the correct ion mobility filtering. Something else must be going wrong, and if we see your files we could probably figure out what that is.
-- Nick |
| |
| aheinone responded: |
2022-08-12 00:13 |
Thanks Brian and Nick. I have Re-imported the raw files always after changing the settings. But maybe it's just some simple setting that I haven't spotted. I also tested different Window types in Ion Mobility filtering, but I don't know, which setting is optimum for our data. I attached the Skyline document here. I uploaded that .raw file into your File Sharing Folder: "220809_Pool1_SureQuant_Survey_500fmol.zip".
Thanks,
Arttu |
|
| |
| Nick Shulman responded: |
2022-08-12 18:01 |
Thank you for sending those files.
Yes, compensation voltage filtering does not seem to be working correctly at all in Skyline 21.2 or Skyline-Daily.
Specifically, whenever there is a spectrum whose compensation voltage does not match the particular precursor, a zero gets added to the chromatogram.
This only happens if there is at least one precursor in your document that does match that compensation voltage. So, if you create a Skyline document where all of the precursors had CV -50, and another Skyline document with only the CV -70 precursors, then the chromatograms in both of those documents would look right. But, this is probably not a feasible workaround for you, especially since some of your peptides have two different charge states, and the two charge states have different compensation voltages.
I am sure that we will figure out how to fix this in Skyline-Daily soon.
-- Nick |
| |
| Brian Pratt responded: |
2022-08-15 14:39 |
Hi Arttu,
Thanks for sending the data, I have identified the problem and the fix will appear in the next Skyline release.
May I have your permission to use this data as part of an automated software quality test? That would make the data (somewhat obscurely) public.
Thanks
Brian |
| |
| aheinone responded: |
2022-08-15 23:50 |
Hi Brian,
Many thanks for you great support! Yes, you can use this data for the software quality tests.
Thanks
Arttu |
| |
| clichti responded: |
2022-09-27 13:26 |
Hi Brian and Nick,
I'm having the same problem as Arttu. We are trying to compare peak areas at two different compensation voltages, and our peak areas are the same with no CV as they are with either of the 2 CVs. Have you been able to resolve this issue yet?
Thanks so much!
Cheryl |
| |
| Brian Pratt responded: |
2022-09-27 14:14 |
Hi Cheryl,
What version of Skyline are you using? The issue we identified with Arttu should be fixed in the current release. Perhaps you just haven't told Skyline what CV to use for each precursor?
Best regards,
Brian Pratt |
| |
| clichti responded: |
2022-09-28 05:38 |
Hi Brian,
I'm using the newest version of Skyline (22.2.0.255). I created an ion mobility library with CVs for each peptide. Within the same data file, I have acquired each of my peptides of interest with two different CVs. If I change the CVs and re-extract, as described above, I get the exact same peak areas as when I do not specify a CV at all. I even tried creating a whole new Skyline file and got the same results. When I manually integrate in the .raw file, I get different peak areas at each CV, as expected. Do I need to try Skyline Daily?
Thanks!!
Cheryl |
| |
| clichti responded: |
2022-09-28 05:52 |
Never mind - I figured out the problem!! It works perfectly if I use "resolving power" as the window type. Thanks so much for all you do! |
| |
| tej nishtala responded: |
2022-12-01 21:31 |
Hi Brian & Nick
As others mentioned above, I observe the same problem with -50CV. I am working on Skyline ver 22.2.0.312
It's a dual CV (-50/-65 CV) dda method on Exploris 480
I was trying to extract Biognosys-11 iRT peptides from the file. I applied the steps decribed in the IMS tutorial with "use results" and was able to extract smooth precursors, however the tranitions aren't the same. Tried the "resolving power" as well, but didn't seem to have an effect on the transitions.
Please find attached screenshot for the ms2 of one of the iRT peptides. The idotp is 0.99 and pfr is 1.0.
Thank you |
|
| |
| Nick Shulman responded: |
2022-12-01 22:11 |
Tej Nishtala,
I cannot tell from your screenshot whether you were expecting it to look different than it does.
If you would like, you could send us your Skyline document and one of your raw files and we can try to figure out what is going on.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including ion mobility libraries and extracted chromatograms.
If that .zip file and your raw file are less than 50MB you can attach them to this support request. You can upload large files here:
https://skyline.ms/files.url
-- Nick |
| |
| tej nishtala responded: |
2022-12-02 16:07 |
Hi Nick
Thanks for getting back to me.
It is not clear as to why the peak shape and boundaries for the transitions are different to the ones without FAIMS.
I'm attaching a screenshot from QE-HF of the same iRT peptide for an earlier experiment.. perhaps that might help in making the distinction.
If not, I can share the files.
Thank you
Best
Tej |
|
| |
| Nick Shulman responded: |
2022-12-02 16:15 |
Tej,
Yes, please send us your Skyline document (or do you maybe have two Skyline documents: one with and one without FAIMS?) and one or more of your raw files. We will figure out what is going on.
-- Nick |
| |
| tej nishtala responded: |
2022-12-05 14:53 |
Hi Nick
Sure, can share the files.
Can you provide an alternate email address to share.
Thank you!
Best
Tej |
| |
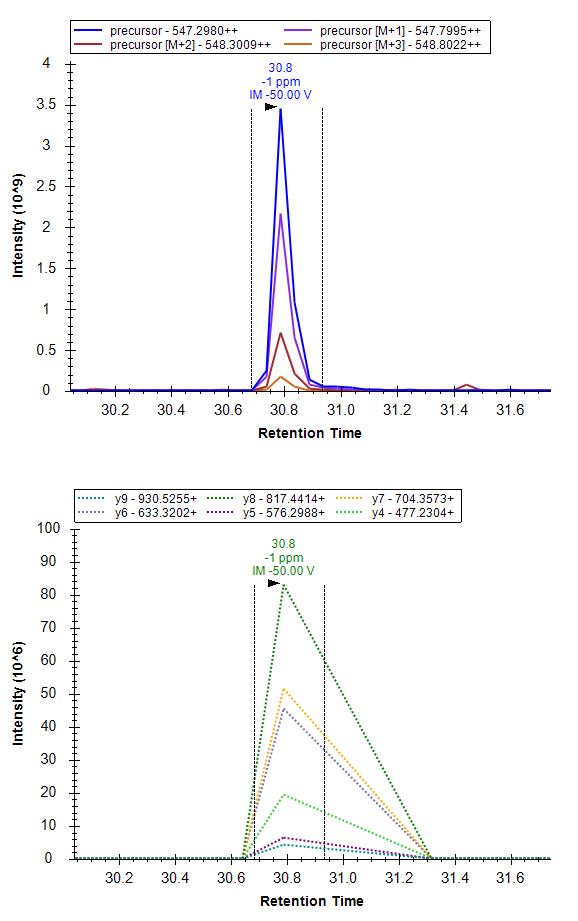 Skyline Exploris 480 dual CV. transitions.jpg
Skyline Exploris 480 dual CV. transitions.jpg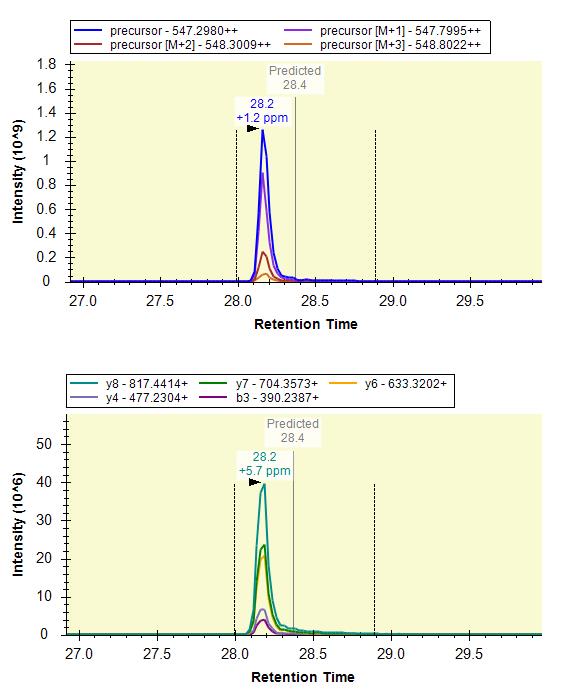 Skyline no FAIMS_QEHF transitions.jpg
Skyline no FAIMS_QEHF transitions.jpg