| Is SkyLine filtering MS/MS peaks with low intensities? | klongnecker | 2022-06-15 04:14 |
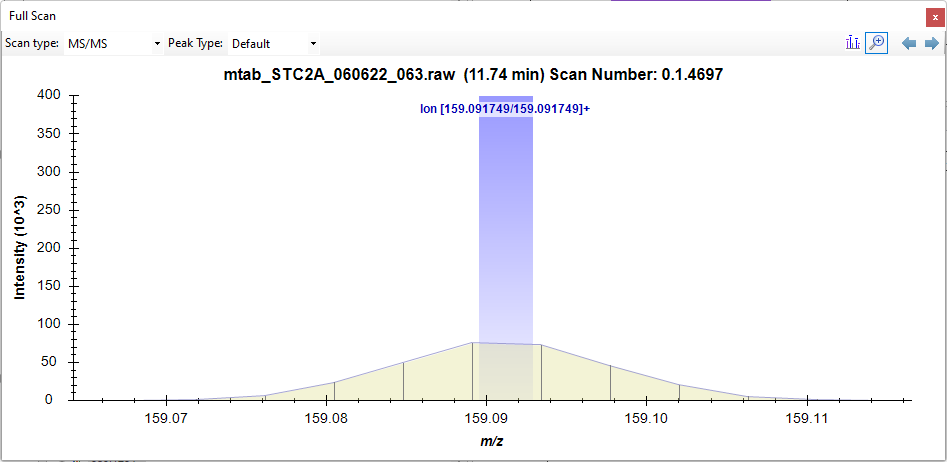
Hello, In a current project I have three sets data for each molecule: unlabeled, 13C labeled, and D5 labeled. I have 13C- and D5-labeled compounds for different standard curves (in the same sample). Hence, this is a use case not within the standard use of SkyLine and I have setup a specific set of molecule names so I can process peak areas that I export out of SkyLine (as raw peak areas, not normalized, no ratios, etc.). We do our quantification on MS1 peaks. However, we use the MS/MS peaks to verify the identity of our known compounds. Hence, even if there is only one MS2 scan with a transition, we find it useful. To make this work I have taken a few extra steps. Thanks to the help you have provided here on the forum (e.g. here, here, and here) I have set the transition settings/full scan to DIA and blanked out the explicit retention times when importing the transition settings. This gets me the MS1/precursors for the unlabeled, 13C labeled, and D5 options. However, I am still missing some transitions. In the PowerPoint file I show an example with tryptophan where I only see one fragment (shown in MZmine and SkyLine) and a second example with tryptamine where I see the two fragments I am expecting. The missing MS2 peak is the lowest intensity, but it still appears in 6+ MS2 scans so I know it is there. Any thoughts on additional settings I may try to have SkyLine show me all (or more) of the MS/MS peaks? Thanks again for your help. One detail - I also uploaded a zip file (from File/Share) that is Longnecker_LowIntensityMS2.zip, and the two *raw files to the FTP site. |
||
 spectrum.png
spectrum.png