| Nick Shulman responded: |
2022-05-26 13:00 |
|
| |
| zzhu248 responded: |
2022-05-26 13:12 |
Hi Nick,
Thank you for your fast response. I have asked one of my lab members and he says it seems like the column "referring" to the path of every self-generated peptide is missing. I am scrutinizing my previous work.
FYI, I have attached the ssl files below. I have tried the one with only last column and it does not work as well |
|
| |
| Nick Shulman responded: |
2022-05-26 13:52 |
Where did you get that .ssl file? Was that a file that you downloaded from supplemental data associated with the paper, or was that a file that you created yourself?
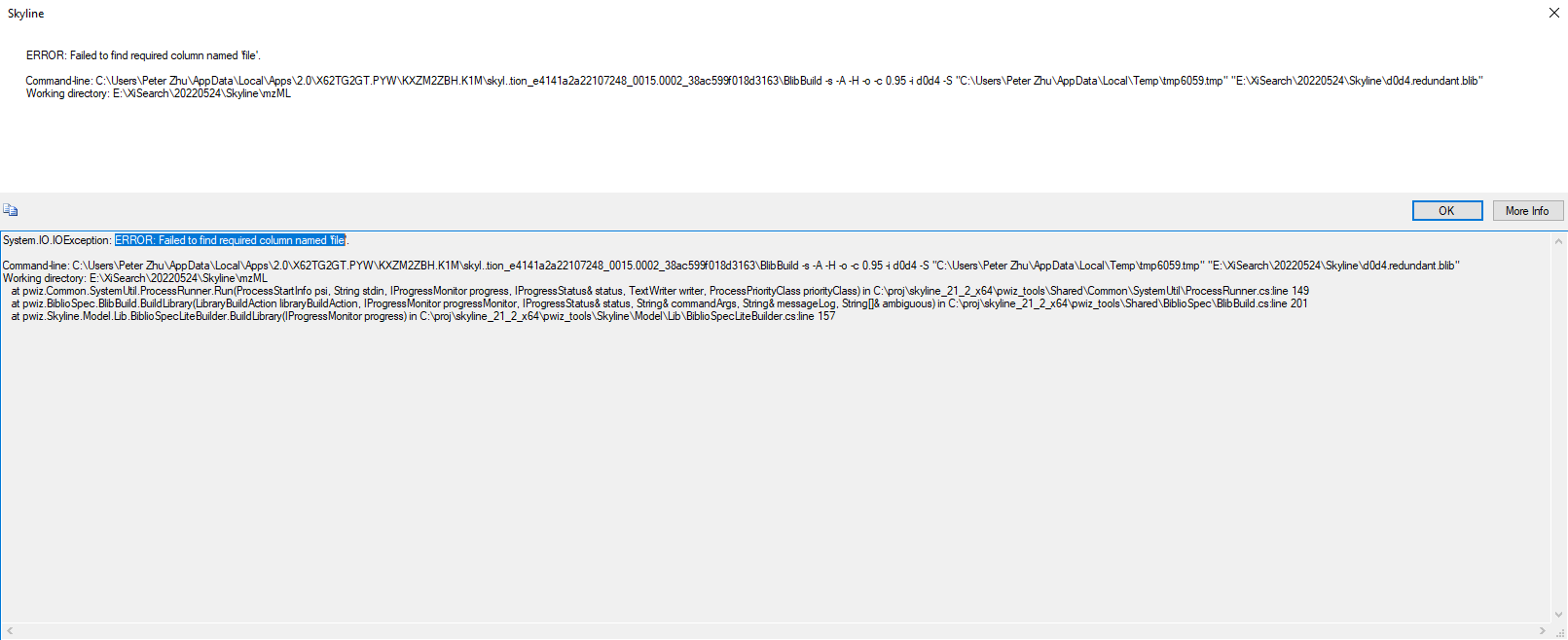
A .ssl file needs to have columns named "File" and "Scan". When BiblioSpec is building a spectral library from a .ssl file, Skyline will look in the raw file referred to in the File column and will find the particular spectrum. That spectrum will be put into the .blib file that is being created.
Is there a reason that you are trying to recreate the steps from this paper? In the last few years, a lot of really good crosslinked peptide search engines have been developed so you should not need to resort to writing a .ssl file with a list of peptides and scan id's.
BiblioSpec can create a spectral library from crosslinked peptide search results so long as those search results have been converted to the "proxl" XML format. On the proxl web page, you can see a list of the crosslinked peptide search engines which can be converted to proxl and can therefore be used by Skyline and BiblioSpec:
https://proxl-ms.org/
-- Nick |
| |
| zzhu248 responded: |
2022-05-26 14:28 |
This is a self-created file. I was applying xiView platform from Rappsilber Lab since they did a very good job on data visualization. I am currently looking forward to a proper way to proceed quantification based on crosslinkers BS3d0 and d4. I have also tried plink and XlinkX and neither of them can provide .ssl file, unfortunately. (TBH, It tooks me 2 days to manually create that file) If you have any suggestions about this field I would be very grateful to your help! |
| |
| Nick Shulman responded: |
2022-05-26 14:44 |
I don't think you should be trying to create .ssl files.
It is possible to convert plink crosslinked peptide search results to proxl, so I think you should do that.
Here is the project page for the tool which converts plink to proxl:
https://github.com/yeastrc/proxl-import-plink2
In the "How to run" section it tells you where to download the file "plink2toProxlXML.jar" and how to use Java to run the jar with commandline arguments to convert a .plink file to proxl.xml
Once you have proxl.xml files you can use those to build a BiblioSpec spectral library.
In Skyline, you can use the "File > Import > Peptide Search" menu item to go through the process of creating a spectral library from proxl search results and then importing proteins from a FASTA file and extracting chromatograms.
Let us know if you get stuck trying to convert your plink search results to proxl.xml.
-- Nick |
| |
| zzhu248 responded: |
2022-05-26 19:39 |
Hi Nick,
I am afraid this does not work for me as it only accepts single linker without performing quantification. I could send them the files directly, but I would still give it a try on my previous method. Anyway, I am very much appreciated for your help and hope you have a wonderful rest of the day.
Best,
Peter |
| |
| Nick Shulman responded: |
2022-10-10 14:03 |
It sounds like things still aren't working for you.
Are you saying that the converter from plink to proxl does not work for you?
Could you send us your .plink files?
-- Nick |
| |
| zzhu248 responded: |
2022-10-10 16:46 |
Hey Nick,
Thank you again for your help. Actually, I found another way (XiSearch) to generate .ssl file and it works. I am currently concerning some of the other trivial issue.
First thing is, though I already have the list of crosslink interactions between groups of proteins, the ssl file itself doesn't contain so I have to manually rematch the linear peptides I inputted to the Skyline with their specific protein crosslinking sites from Xisearch or plink result. From this morning tutorial, I am wondering if we may have automatic strategy to align them and grouped by the proteins they attached, just like our PRM practice.
(P.S. Like the last slide of your CrosslinkingInSkyline.ppt, it only gives space for 1 protein but I usually focus on interaction between different proteins)
The second one I just sent a email to Mark this morning. The problem I am facing is from a quantitative crosslinking project whereas control group and experimental group were labelled by light and heavy labeled crosslinker in 1:1 mixture in forward and reverse way (each has 2 replicates). Here, the challenge is for the same target peptide from 2 separated spectral libraries, I am usually facing nice signals in forward labeled set but chaotic in reverse labeled (though I was expecting peak intensity difference are switched by 2 groups). If you click on the reversed one, the previous forward set idotp values becomes crazy. I have attached the example peak areas below.
Best,
Zexin |
|
| |
| Nick Shulman responded: |
2022-10-10 16:55 |
Can you send us your Skyline document?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including spectral libraries and extracted chromatograms.
If that .zip file is less than 50MB you can attach it to this support request.
You can upload larger files here:
https://skyline.ms/files.url
I was not able to read the text on the screenshots that you attached because the resolution was too low but if I see your Skyline document I will probably be able to figure out what you were looking at.
-- Nick |
| |
| zzhu248 responded: |
2022-10-10 17:10 |
Here we go. Thanks again for your fast response and guidance. |
| |
| Nick Shulman responded: |
2022-10-10 17:33 |
Thank you for sending that Skyline document.
Your document does not contain any crosslinked peptides. There are some modifications defined which should be crosslinkers ("BS3 crosslink", "Xlink:BS3-link", "Xlink:BS3d4-link", "BS3D4 crosslink"), but the peptides that are using them do not have them attached to anything.
Can you give an example of a crosslinked peptide that you would like to have in your document? That might be two peptide sequences that are attached to each other with a crosslinker, or it could be one peptide sequence with a crosslinker connected two amino acids within that peptide sequence.
After you tell me a crosslinked peptide that you would like to have in your document, I can walk you through how to add it.
The two ways of getting a crosslinked peptide into your document are using the "Edit Modifications" dialog on page 3, or using the special Modified Sequence syntax on pages 9 and ten of the Crosslinking pdf:
https://skyline.ms/wiki/home/software/Skyline/download.view?entityId=ac4c6cc4-97c6-1038-8aae-e465a393ee52&name=CrosslinkingInSkyline21_1.pdf
-- Nick |
| |
| zzhu248 responded: |
2022-10-10 18:11 |
Hmmm actually I am following based on the protocol of this paper. What they did is they merge crosslinked peptides by adding pseudo-residue between them then recognized by Skyline. (I used K+26 for BS3 and K+30 for BS3D4 cuz this is what ssl file derived.)
Here are the crosslinked csv data as well. I am not sure if it is because the version updates or other reasons, some of the screenshots from the ppt are somehow different from the description in the paper. |
|
| |
| Nick Shulman responded: |
2022-10-10 18:36 |
The crosslinked peptide feature was added to Skyline in 2020. That paper was written before Skyline understood anything about crosslinked peptides, so they had to pretend that their crosslinked peptides were something else.
Would you like to reproduce the behavior of Skyline that these researches faced when they did their experiments 3 years ago, or would you like to take advantage of the new Skyline crosslinking features?
Your Skyline document has a lot of variable modifications defined. Skyline wants to give you all of the possible permutations of those variable modifications, so you have a lot more peptides in the Targets tree than you probably want. You should go to:
Settings > Peptide Settings > Modifications
and remove some of the modifications which are identical to each other.
-- Nick |
| |
| zzhu248 responded: |
2022-10-10 19:45 |
Definitely prepared for something new! No wonder I have spent such a long time on this......
So back to the beginning. I actually trusted my plink2 result more, but the developer previously said it does not support multiple crosslinkers. I tried once but failed unfortunately. I sent them the email, but no one replied. I just attached plink file here.
By the way, if you feel zoom meeting is more convenient and faster than forum, we may schedule a time base on your availability. I would be extremely appreciated to your guidance.
Best,
Peter |
|
| |
| Nick Shulman responded: |
2022-10-10 20:14 |
It certainly looks like the latest version of plink would support more than one crosslinker.
If you look at page 10 of their documentation there is a dialog which lets you select multiple crosslinkers:
http://pfind.org/software/pLink/pLink2%20User%20Guide.pdf
Unfortunately, I do not know how to use plink.
The complete list of crosslinking search engines which can be made to work with Skyline (because their search results can be converted to .proxl) can be found on this page:
https://proxl-ms.org/
-- Nick |
| |
| zzhu248 responded: |
2022-10-10 20:37 |
I know. plink 2 is absolutely fine. It's always the transfer step. Let me retry this proxl website again.
Thanks again and see you later this week.
Best,
Zexin |
| |
| zzhu248 responded: |
2022-10-10 21:02 |
It is reported "could not find the main class". |
| |
| Nick Shulman responded: |
2022-10-10 21:26 |
"Could not find the main class" is an error that Java gives you if you make a mistake in telling it what to run.
The way to run the plink converter is:
java -jar plink2toProxlXML.jar
It is important to have the "-jar" in there because that tells java to look inside the file "plink2toProxlXML.jar" and figure out what the name of the main class is.
If you leave out the "-jar" the Java thinks that "plink2toProxlXML.jar" is the name of the main class (and also, java has no idea where to find the definition of that main class because you did not tell it any jar files to look in).
Is "Could not find the main class" the actual error you are seeing?
Did you try running java with a commandline, or did you double-click on something in Windows explorer?
-- Nick |
| |
| zzhu248 responded: |
2022-10-10 21:40 |
I tried both. I have to apologize I am a rookie in java. The command prompt says "missing required options [--param=<paramFile>, ---out=<outFile>, -- fasta<fastaFile>]. Though I have already put them under same folder. |
| |
| Nick Shulman responded: |
2022-10-10 22:29 |
If you put "plink2toProxlXML.jar" in the same folder as your .plink file, then the commandline would be something like:
java -jar plink2toProxlXML.jar -p "pLink_task_2022.05.23.13.14.06_did0+citd4.plink" -r . -o citd4.proxl.xml -f mouse.fasta -b C:\pFindStudio\pLink\2.3.11\bin
The thing after the "-f" should be the path to a FASTA file. I don't think it matters what FASTA file you choose, because it will not affect any of the data that Skyline cares about. The thing after "-b" should be the path to where plink was installed on your computer. The thing after "-o" can be any filename which ends in ".proxl.xml" (it's the file that will get created as a result of this process)
Unfortunately, a few things seem to be going wrong:
1. When I run this with the plink2toProxlXML.jar that I downloaded from the proxl-import-plink2 website, I get an error "Encountered an error during conversion: javax/xml/bind/JAXBContext"
2. I was able to get the proxl converter to run using a debugger, but the .proxl file cannot be read by BiblioSpec (get an error "only Byonic or Percolator ProxlXML files are supported;")
I will email the developers of Proxl and BiblioSpec and get an estimate of how hard it will be for them to get this scenario working.
-- Nick |
| |
| Matt Chambers responded: |
2022-10-11 11:55 |
Hi Zexin,
The next version of Skyline-daily should have BiblioSpec fixed to support pLink proxl XML files. Nick's instructions here should get you going with converting from pLink to proxl XML with the Java converter. The converter author told us the error Nick mentioned about JAXBContext is probably due to running it with something newer than Java 8. So you may have to download that or wait for the converter developer to release a new version with that problem fixed.
-Matt |
| |
| zzhu248 responded: |
2022-10-11 12:27 |
Hey Matt,
Thanks for your message. The java version on my computer is java 8.
Do you have an estimation time when our new version of skyline-daily will be available? The converter developer has not updated it since 2020 and he didn't reply my message previously. Hope he can provide the update soon as well.
--Zexin |
| |
| zzhu248 responded: |
2022-11-09 09:57 |
Hmmm it seems like the latest patch does not include the fix that .proxl file read by BiblioSpec. (PS. I have derived proxl.xml file via that java tool)
Best,
Zexin |
| |
| Nick Shulman responded: |
2022-11-09 10:14 |
Make sure that you are using the latest Skyline-Daily.
A patch for regular Skyline 22.2 was released yesterday, but that would not be expected to have this fix in it.
You would need to use Skyline-Daily. There was a Skyline-Daily update on November 2nd, and I would expect this bug to be fixed in that.
If this isn't working for you with the latest Skyline-Daily then it would be helpful if you could send us your .proxl.xml file.
-- Nick |
| |
| zzhu248 responded: |
2022-11-09 14:26 |
Hey Nick,
I retried it on the Skyline-Daily, but it asks for the mgf/mzML file. But actually in the plink, I merged several replicates raw data in one run. It seems like Skyline does not recognize the subtitle of mgf file of every single replicates'that plink derived.
I have attached my file below hopefully you may understand.
Thank you again for your precious time and help.
Best,
Zexin |
|
| |
| Nick Shulman responded: |
2022-11-09 16:30 |
Do you have a file called maybe "Cit_did0+citd4XL1.mzML" or "Cit_did0+citd4XL1.mgf" or something?
When I try to create a spectral library from your proxl.xml file, BiblioSpec tells me that it's looking for a file whose name starts with "Cit_did0+citd4XL1" and which has one of a number of different possible extensions.
If you have a file with that name, could you send it to me?
Is this the same error that you are seeing?
If you're seeing a different error, could you send me a picture of the error that you are seeing?
If there is no file called anything like "Cit_did0+citd4XL1", then you might have to send me all of the files that were in the same folder as your .plink file, and I can try to find someone to figure out why we end up thinking the spectra will be contained in a file whose name starts with ""Cit_did0+citd4XL1".
If you have to send us a large number of files, you can package them in a .zip file and upload them here:
https://skyline.ms/files.url
-- Nick |
|
| |
| zzhu248 responded: |
2022-11-10 09:04 |
Yes, it is the same error, and that's the most intriguing part. It doesn't have such a file naming like this.
The .mgf file that plink created is called "Cit_did0+citd4XL1_HCDFT.mgf". But even if I renamed it, it didn't work yesterday. Therefore, I was suspecting that the .XML file is derived by "Cit_did0+citd4XL1" and Cit_did0+citd4XL2" for multiple replicates.
Fortunately, this morning I regenerated the .mzML file via MSConverter for those 2 and it works. However, when I am adding the peptides in the library to the target list, it automatically includes the linear peptides. Is there any way that I may modify the settings for the filter that only crosslinked peptides will be included in the list?
Best,
Zexin |
|
| |
| Nick Shulman responded: |
2022-11-10 09:36 |
I cannot think of a way to use the settings to only give you peptides that have a crosslink in them.
However, after you have everything in the document, you can use the Document Grid to delete all of the peptides whose Modified Sequence does not have a crosslinker in it.
In the Document Grid, you would choose "Peptides" from the Reports dropdown and then right-click on the "Peptide Modified Sequence" column and choose "Does not contain" and specify the name of the crosslinker.
After the filter has been applied you can select all of the rows and then use either the red X on the Document Grid toolbar or the "Actions" dropdown to delete all of the selected peptides.
If you would like you learn more about the Document Grid you should look at the Custom Reports tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_custom_reports
-- Nick |
| |
 Screenshot 2022-05-26 143119.png
Screenshot 2022-05-26 143119.png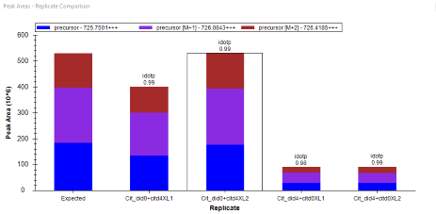 1.png
1.png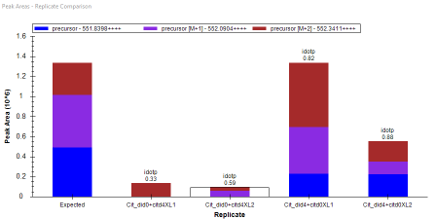 2.png
2.png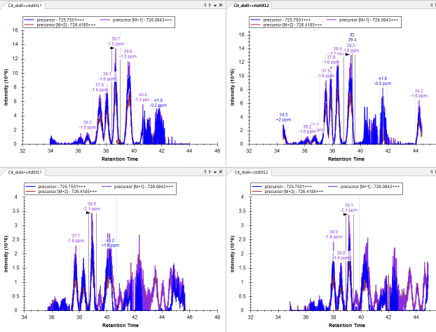 3.png
3.png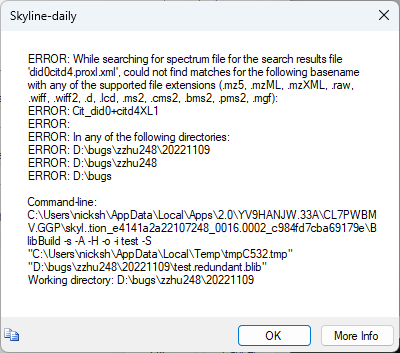 MissingSpectrumFile.png
MissingSpectrumFile.png Screenshot 2022-11-10 110311.png
Screenshot 2022-11-10 110311.png