Hi All,
I am looking at plasma data from DIA study
Forchelet et al., 2018 done on a SCIEX 5600. The authors did not create their own library, but rather used a publicly available SWATH assay library
Liu et al., 2015; PXD001064 2018 also generated from plasma.
The Liu et al., library lacks my protein of interest, which means it was never identified in the Forchelet dataset.
As a result, I searched and found a dataset on ProteomeXchange
Hondius et al., 2021; PXD023199 that also used a QTOF 5600. While these samples were from human brain, the data contained my protein of interest and was an instrument match.
I pulled the DDA data and repeated their database search steps in MetaMorpheus as described in the paper.
Using the “Import DIA Peptide Search” wizard, I created a spectral library with the Hondius search results and spectral files. At the add chromatograms step, I uploaded the Forchelet .wiff files.
Please see PPT attached or full workflow details.
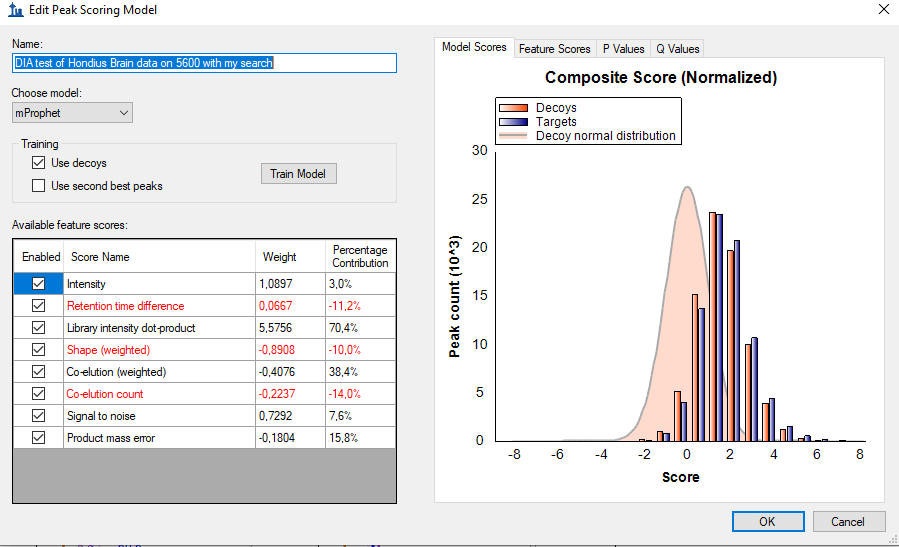
The issue I am having is that the Decoys and the Targets perfectly overlap with each other. So, I cannot tell what is real and what is not. It is not the ideal situation, but I have to work with what I have since the library in the Forchelet study had none of my proteins of interest so they could never be found even if there were there.
I could always try this approach by
Searle et al., 2020: Gerating high quality libraries for DIA MS with empirically corrected peptide predictions;t but the issue would be the same as I am not generating DDA data with the same gradient, instrument (in GPF), or time span.
Advice much appreciated.
Norelle
 Capture.PNG
Capture.PNG