I am working with low intensity peptides in PRM and try to get as much evidence as possible.
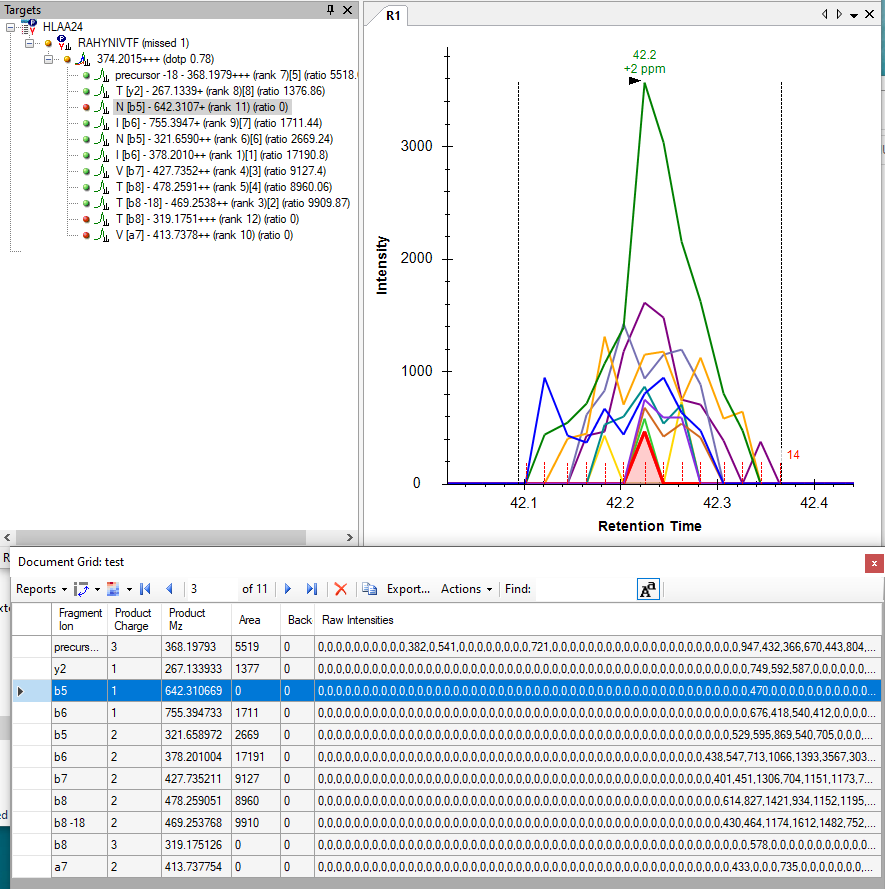
Recently I came across a peptide that had some transitions always marked in red (area 0) even though there is some signal.
I tried to force reintegration or reimporting the data and had no success.
I am attaching an image showing this should not be related to the background subtraction.
Maybe you can tell right away what is up.
When Skyline is detecting and integrating peaks, the first thing that Skyline does is interpolate the chromatogram in the time dimension so that Skyline has a set of intensities with evenly spaced times.
You can see what that interpolated chromatogram looks like by right-clicking on the chromatogram and choosing "Transform > Interpolate".
One thing that often happens during that interpolation process is that chromatograms which have only one non-zero intensity like that end up turning into a completely flat chromatogram.
When chromatograms are extremely jagged like this, the interpolation algorithm that Skyline uses can have a profound effect on the peak areas that Skyline reports.
-- Nick
jfoe responded:
2022-01-26 09:14
Thanks for the nice explanation.
I never noticed it like this even though I work on chromatograms like that a lot.
I suppose there is no way to play around with that setting? I'd like to better understand how this tends to affect my dotp results.
Or maybe some resource to understand how this interpolation works exactly? It doesn't seem straight forward to me.
There are no settings that would adjust the way that Skyline does this interpolation.
A few years ago, I did implement a different interpolation algorithm which would guarantee that it would not change integrated peak areas, but that new algorithm was never made part of Skyline because it ran slower than what we already had, and, so far, no one has said it was important that we change this behavior.
-- Nick
jfoe responded:
2022-01-27 02:48
Dear Nick,
thanks for all the details!
The way I read this is that the interpolation is probably helpful in many cases but an option to just integrate the intensities over time as is would be reasonable to include I think.
So if the chance presents itself again I'd be a voice in favor for sure!
I am still puzzled that I never noticed this before so just to make sure:
Is this in any way related to the new MS/MS acquisition method settings? I now use the PRM setting whereas I used to use the targeted setting, which is now deprecated it seems.
I much appreciate all that skyline helps with my work.
 peak_area_issue.png
peak_area_issue.png