| PTM scoring | dkueltz | 2022-01-02 11:37 |
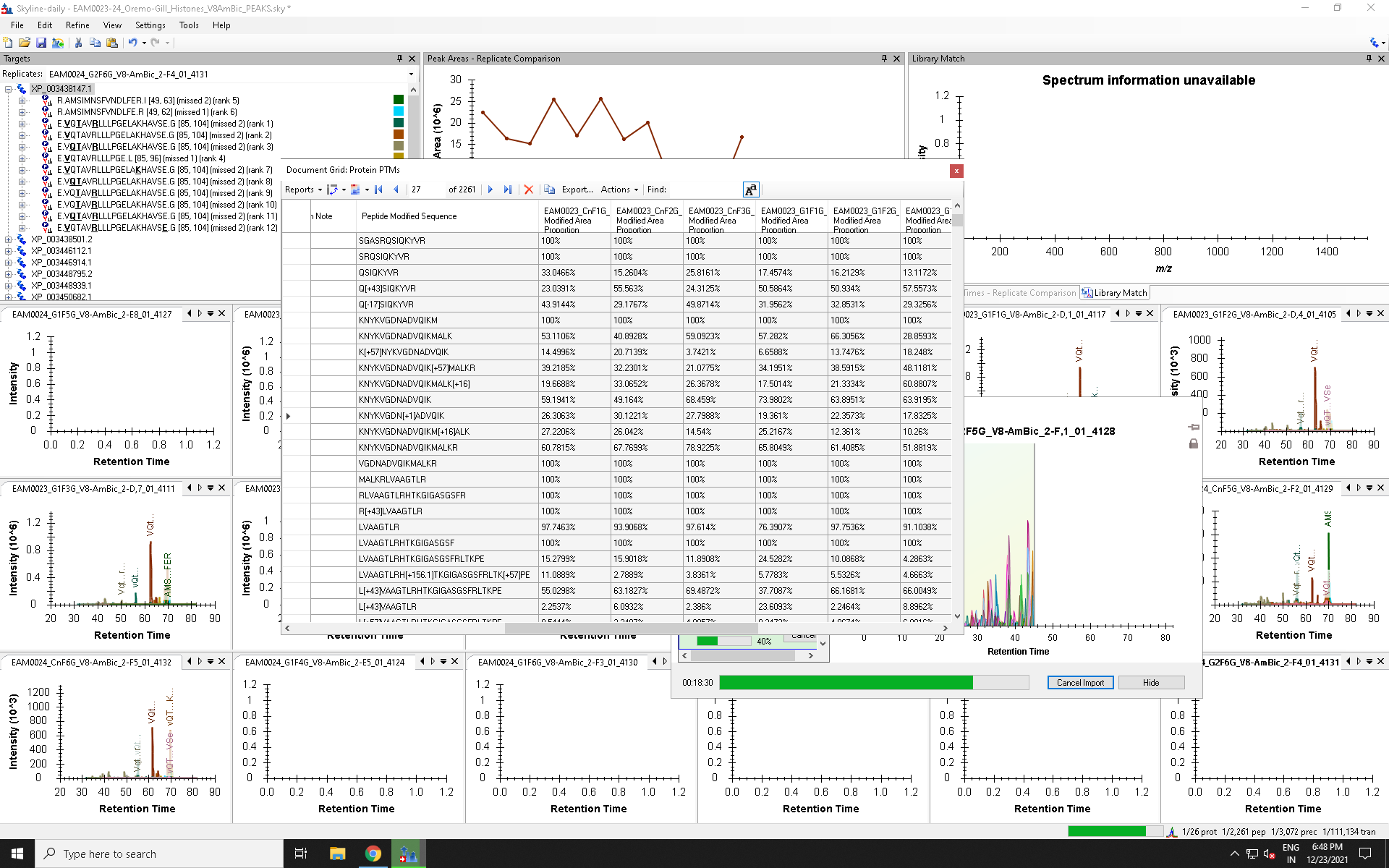
Hi Nick, Brendan et al., The following is a wishlist item I have for future implementation in Skyline re: PTM scoring. Right now the scoring of PTM peptides is based on the ratio of modified versus unmodified peptides but that ratio is based on the peptide sequence rather than the AA position. This works fine if one does not have any missed cleavages or semi-cleaved peptides but in reality there is always a certain amount of missed cleavages and semi-cleavages in complex samples or histone extracts collected from cells or tissues. It would be more accurate to base the ratio of modified versus unmodified peptide on the AA position in addition to the type of modification such that peptides of different starting and ending AAs (semi- or missed cleavages) that contain the PTM AA will be considered for calculating the overall peptide ratio. So the PTM ratio would be the ratio of the sum of transition intensities for all peptides that have a specific PTM in a specific AA position over the sum of transition intensities for all peptides that include that AA position (whether modified or not). This ratio would not require normalization as it is intrinsic to each sample and it would allow relative comparisons of PTM ratios from samples collected under different biological contexts. My lab is currently calculating these ratios by exporting raw transition areas from SKyline and generating a corresponding excel template but generating this template manually takes a very long time. I think this would be a very nice function to include in Skyline that should be of interest to anyone working on PTM quantitation. I am attaching a screenshot that illustrates that the ModifiedAreaProportion is currently calculated only for exactly the same peptide. However it should be calculated for any peptide that includes the modified AA even if the peptides differ in start/ end/ length. Otherwise PTM ratios will be inaccurate. For example in the screenshot the peptide KNYKVGDNADVQIKM (unmodified) is shown as having a 100% value even though there are other peptides (of different start or end AA) that are modified at AAs included in that peptide. So basing these ratios on identical peptide seqs rather than identical AA position gives misleading results re: peptide PTM ratio. Thanks for considering this wishlist request, |
||
 ModifiedAreaProportion.png
ModifiedAreaProportion.png