Dear Skyline team,
Hello, my name is Hogeun.
I am testing the prm-PASEF using skyline-daily(21.1.9.335).
We took our PASEF data including Ion Mobility using timsTOF Pro 2.
And the characterized DDA file was extracted from PEAKS (Xpro).
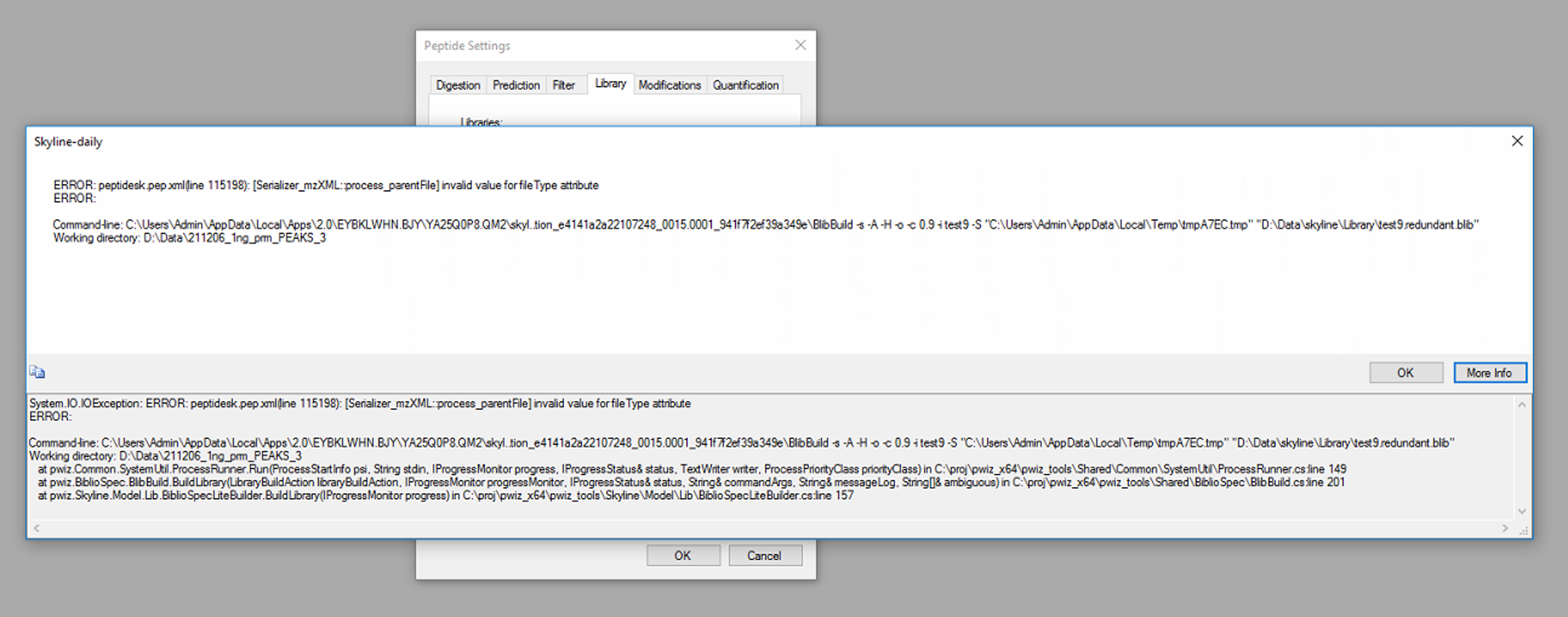
I found the attached message when I input the .pep.xml (including IM data from PEAKS) in library option in Peptide setting .
Do you know the reason? Could you give me the solution if you know that?
best regards,
Hogeun Kwak
 Error.png
Error.png