| Full-MS/ddMS2 data import | ingus perkons | 2021-11-08 14:08 | |||||||||||||||||||||||||||||||||||||||||||||||||
Hello, However, I wanted to process some Full-MS/ddMS2 data acquired on Orbitrap using an inclusion list. The problem I encounter: I cannot extract (see) the corresponding ddMS2 product ion scans. The data files are not corrupt since I can extract ddMS2 scans in both XCalibur and MZmine and there are no import errors whatsoever in the Skyline itself. I suspect the problem is either with (a) Transition list or (b) Transition settings. I have attached an example data file that I want to process (" Example_Full-MS-ddMS2.mzML" ) and corresponding screenshots from the imported Transition list (Capture_a.png), Transition settings (Capture_b.png) + the screenshot of what I see once everything is imported and extracted (Capture_C.png). _I have enabled View --> Transitions --> All. _ Interestingly, when I set the mass accuracy settings in " Transition settings --> Full-scan" to 100 ppm in both “MS1 filtering” and “MS/MS filtering” panels, I can see ddMS2 scans very well (see "Capture_D.PNG"). Yet, if I set 10 ppm in “MS1 filtering” and 100 ppm in “MS/MS filtering” or vice versa - ddMS2 data again disappears after re-import. I'm puzzled because the raw data are of good quality and the mass errors are well below 10 ppm (see Capture_E.PNG). Tried to find the answer in tutorials and the forum, but, apparently, could not notice it. |
|||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
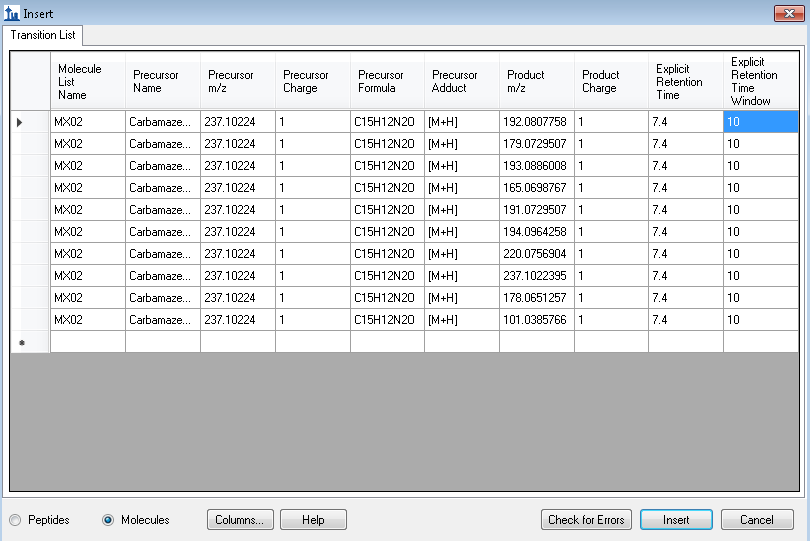 Capture_A.PNG
Capture_A.PNG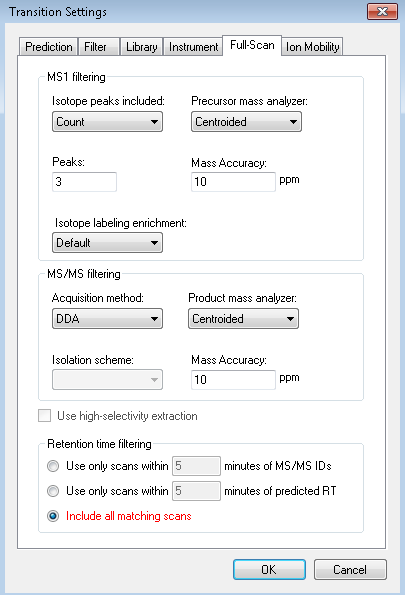 Capture_B.PNG
Capture_B.PNG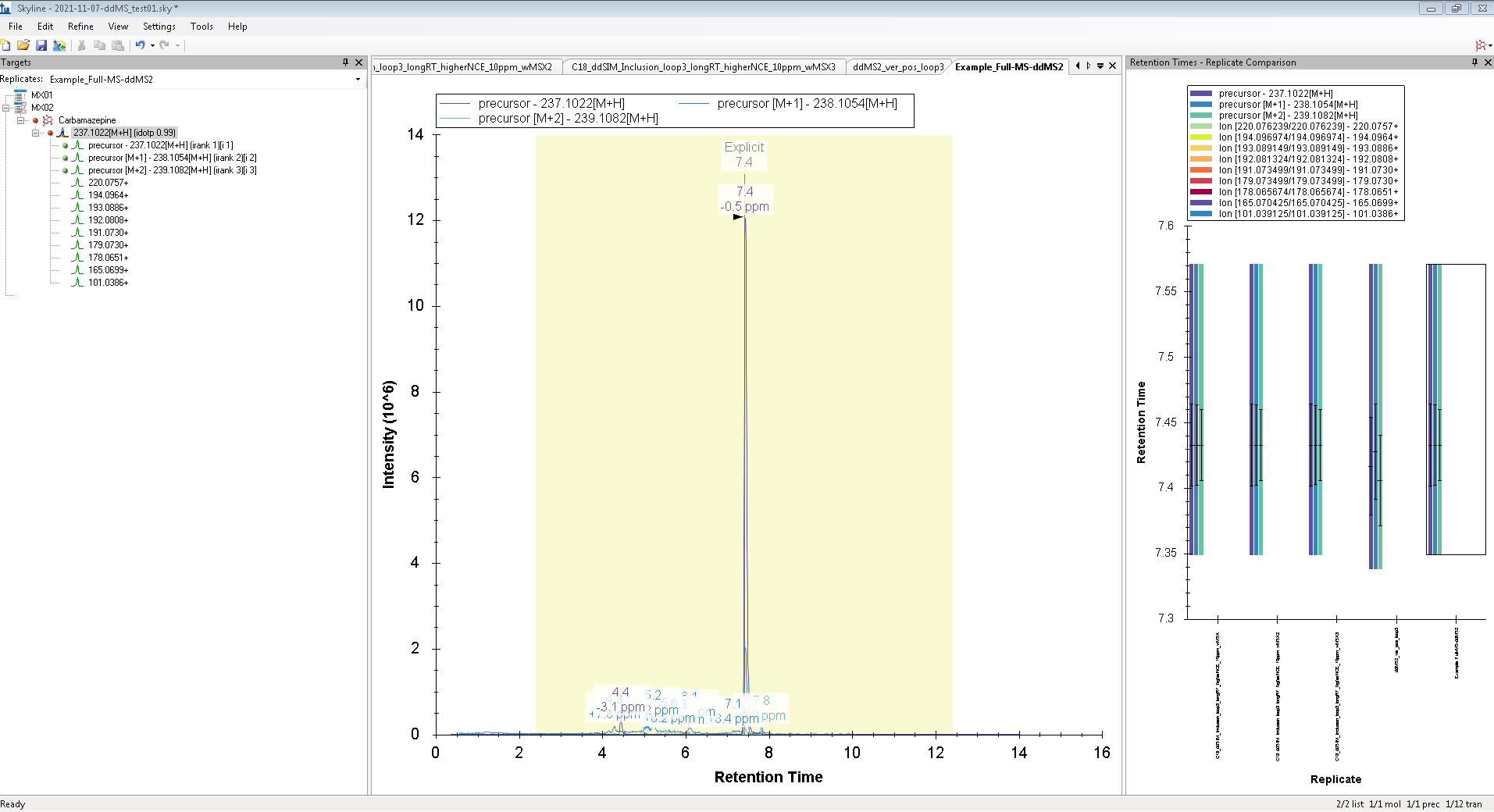 Capture_C.PNG
Capture_C.PNG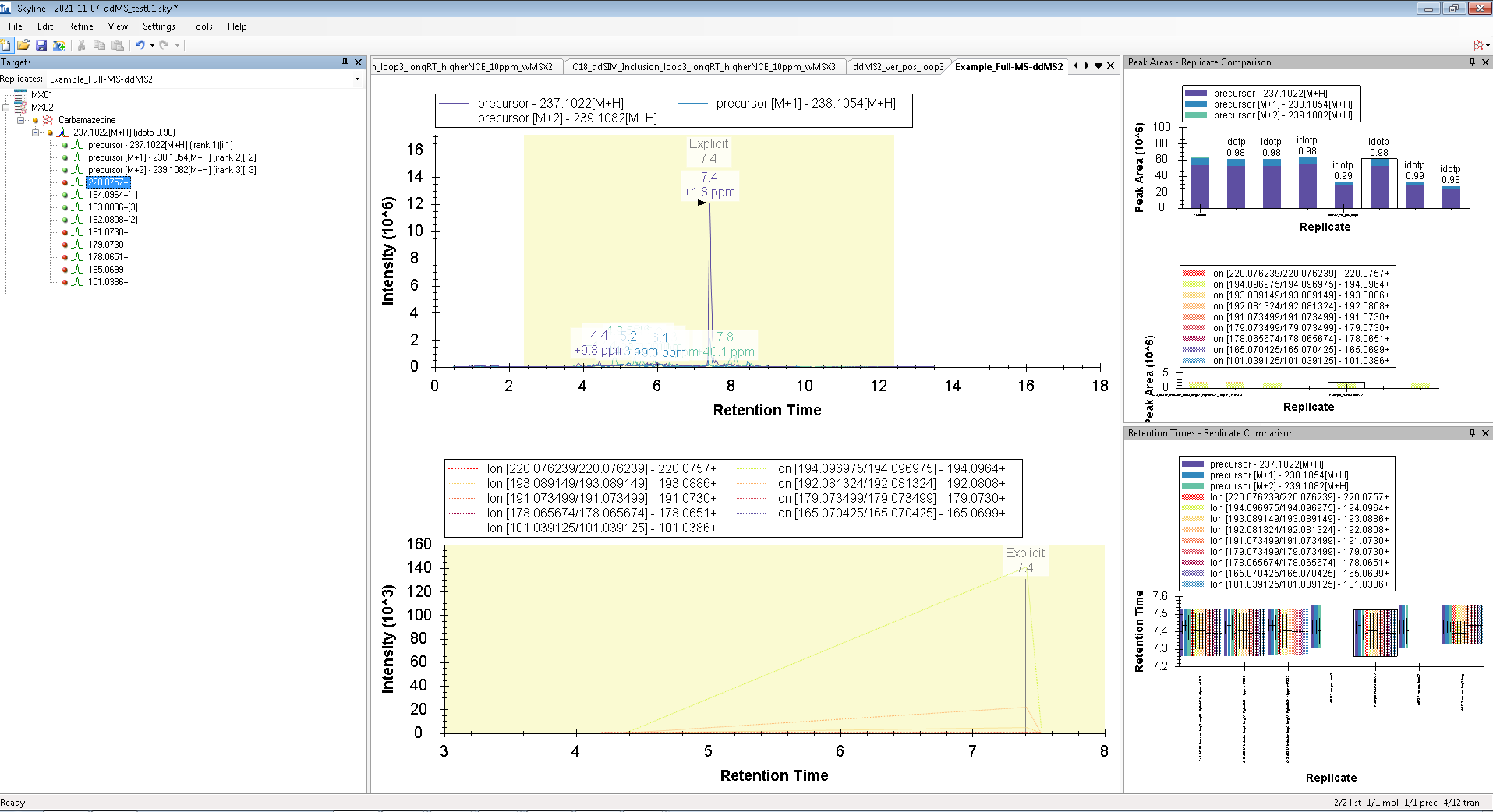 Capture_D.PNG
Capture_D.PNG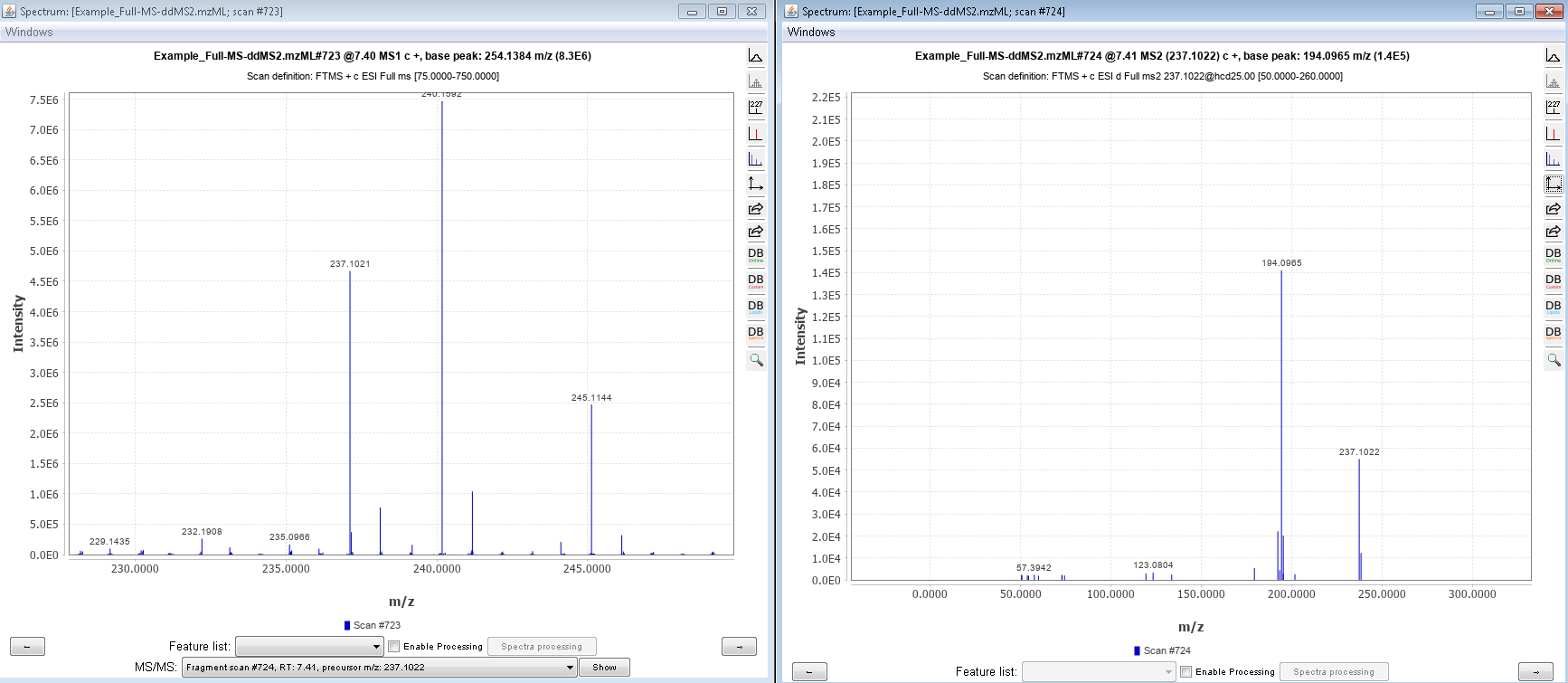 Capture_E.PNG
Capture_E.PNG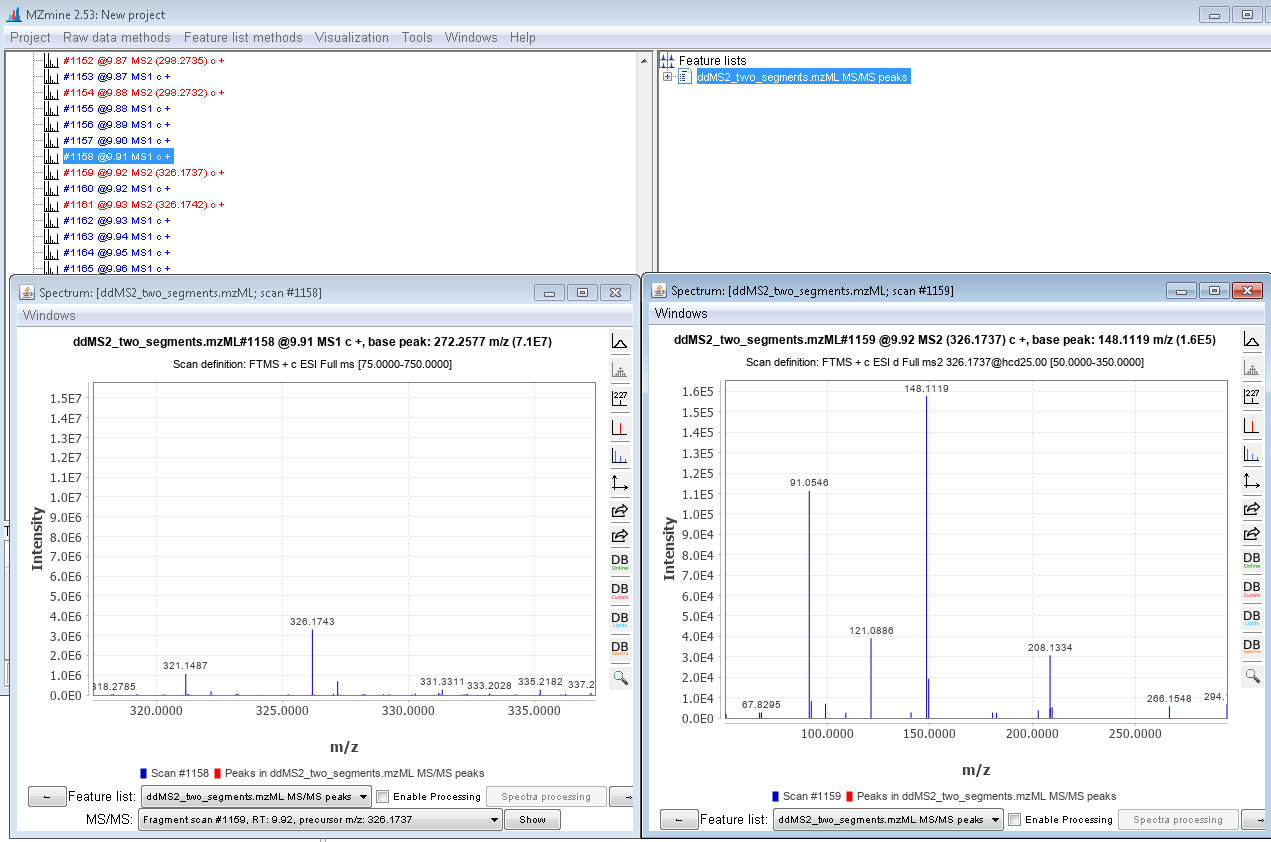 Capture_F.PNG
Capture_F.PNG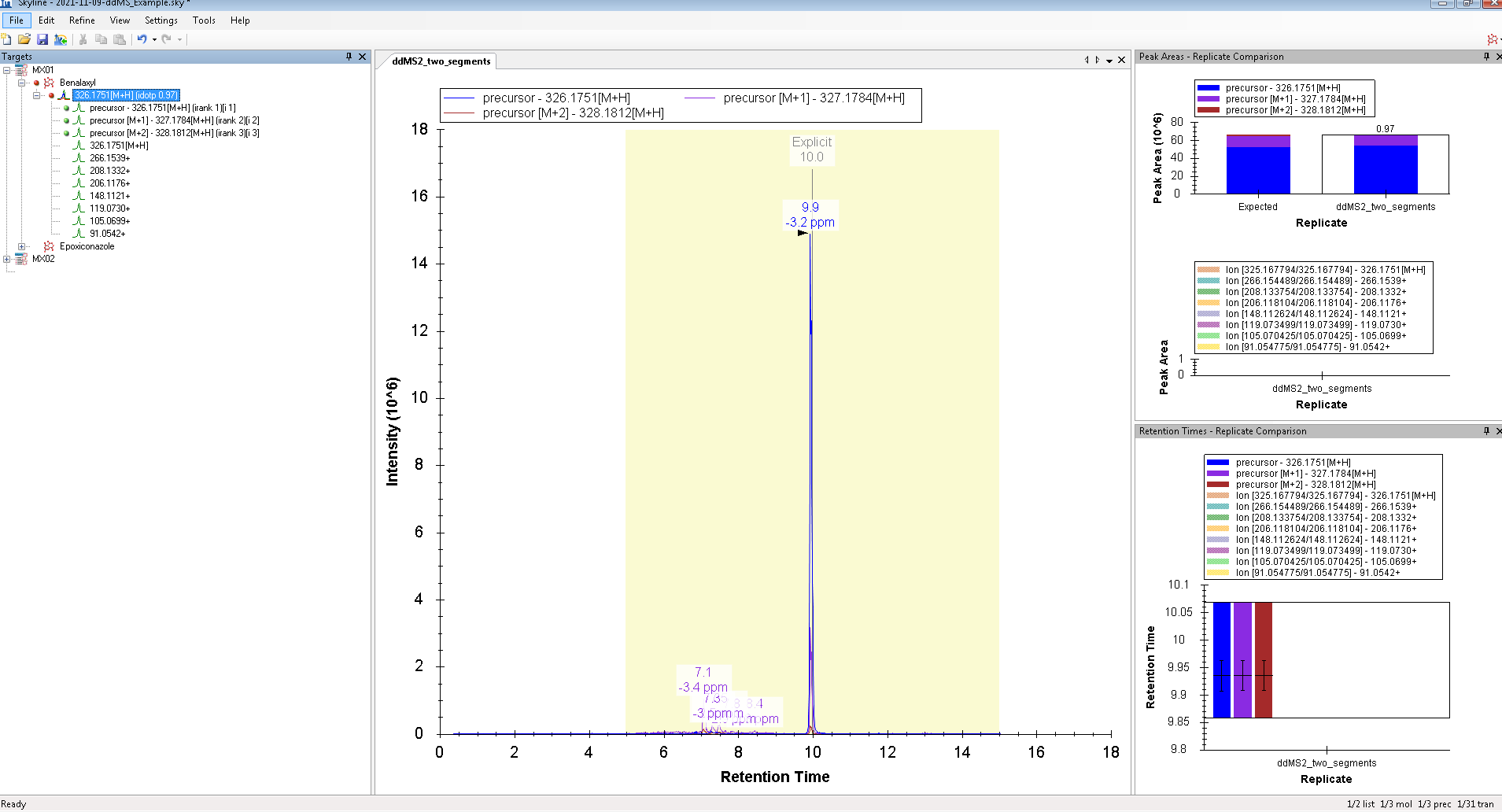 Capture_G.PNG
Capture_G.PNG