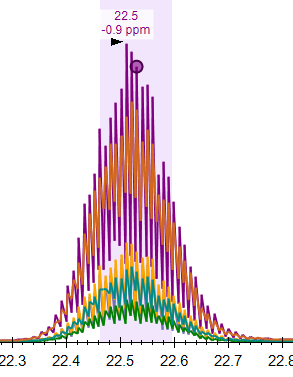
When your chromatogram looks jagged like that, it's often because there are two different sorts of spectra that are contributing the chromatogram.
In a PRM method, the problem might be that there are two different isolation windows which the mass spectrometer targeted which both contain the precursor m/z. Then, you end up with a high intensity point on the chromatogram corresponding to the spectra which were targeting your precursor, and slightly lower intensity points that come from the spectra which were targeting a slightly different m/z. The way that you deal with something like that is to change the isolation scheme that you have specified at "Settings > Transition Settings > Full Scan" so that Skyline only thinks your precursor can be found in one sort of spectra.
A different thing that might happen is that you told the mass spectrometer to collect two different sorts of spectra. Maybe some of the spectra are low resolution, and some are high resolution, or some spectra come from the orbitrap and some come from the ion trap, and you were hoping that Skyline would only use one set of the spectra for the chromatogram extraction. If something like that is the case, the best thing to do is to use ProteoWizard MSConvert to create a .mzML file which is missing the spectra that you do not want Skyline to see.
If you send us your Skyline document and raw file, we might be able to figure out what is going wrong and give you better suggestions.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
You should send us that .zip file as well as at least one of your raw files.
Files which are less than 50MB can be attached to this support request.
You can upload larger files here:
https://skyline.ms/files.url
-- Nick
 skyline_question.PNG
skyline_question.PNG