| Difficulties reading SWATH results | laura corveleyn | 2021-09-27 02:14 | |||||||||||||||||||||||||||||||||||||||||
Hi, I am having some troubles when importing SWATH data as results. I want to compare histone peptidoform expression across several conditions (cell lines) using peak areas. To do this, I made a QC sample (mixture of all conditions) as a baseline. However, when importing the SWATH raw data of these QC samples into Skyline, it seems like something goes wrong when reading it. The XICs look a bit strange. I tried reimporting results several times and made a new Skyline project, but nothing helps. You can find a screenshot of some XICs in the attachment (the QC samples are in the bottom left). Hope you can help me out :) Thanks in advance! Kind regards, Laura |
|||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||
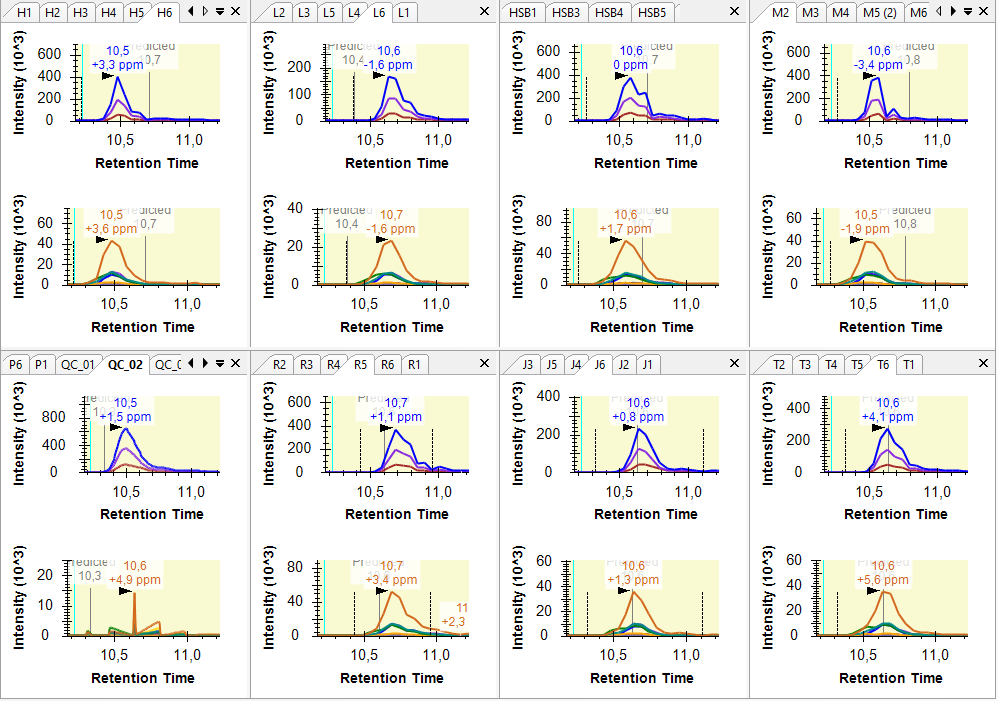 XIC_QC.PNG
XIC_QC.PNG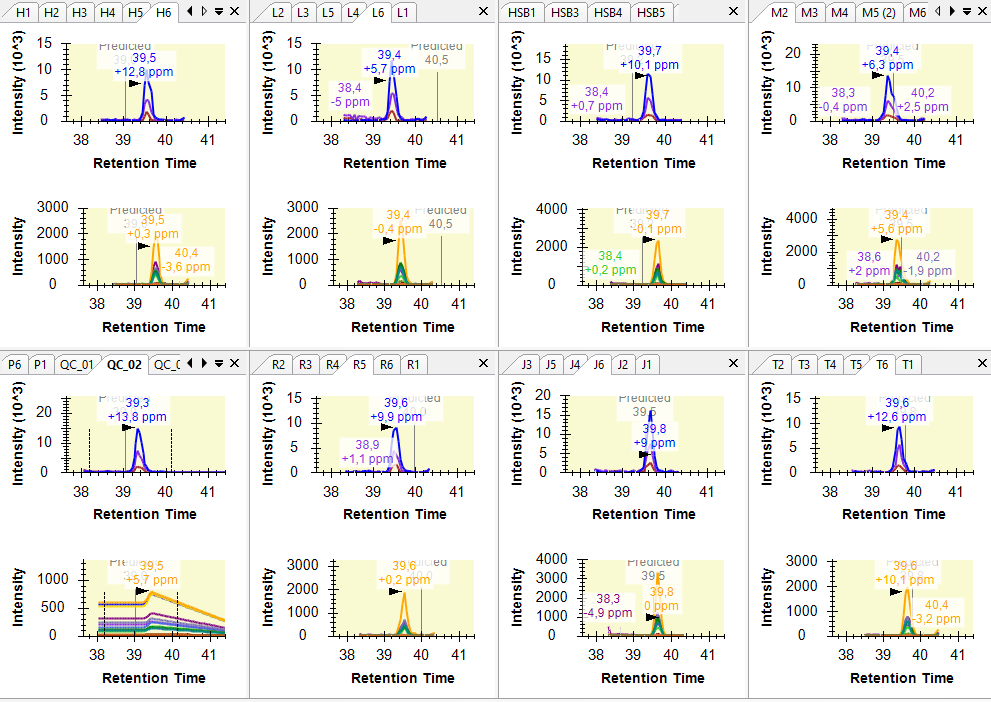 XIC_QC_2.PNG
XIC_QC_2.PNG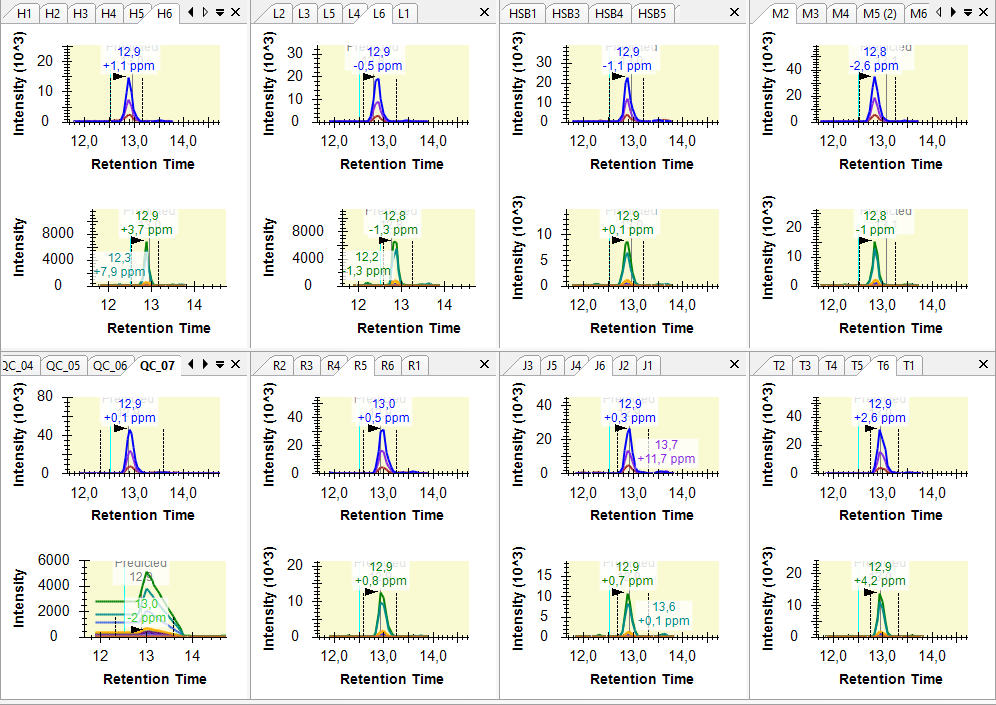 XIC_QC_3.PNG
XIC_QC_3.PNG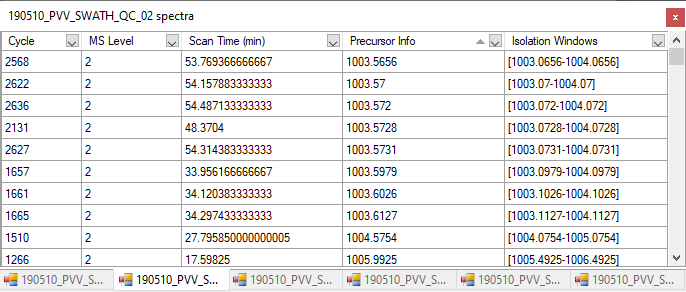 SpectraSortedByPrecursor.png
SpectraSortedByPrecursor.png